X = [Mg, Fe2+, Mn2+]2−x[Fe3+, Al, (Mn3+, Cr3+, V3+)]x;
Y = Fe3+, Al, (Mn3+, Cr3+, V3+, Ti);
Z = Si
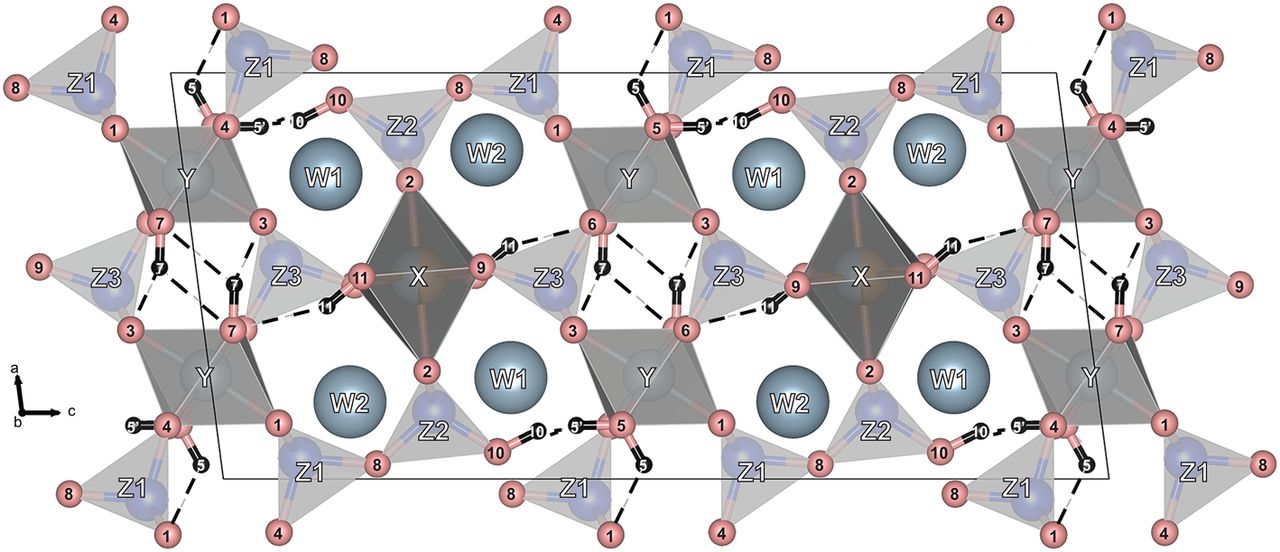
Pumpellyite is crystallographically related to epidote with two independent chains of edge sharing octahedra (Yoshiasa & Matsumoto 1985; Nagashima et al. 2010). Pumpellyite is commonly idealized as containing divalent cations at half of the X sites (Coombs et al. 1976 ; Nagashima et al. 2010). Pumpellyite contains four hydroxyl sites if X contains only divalent cations, but only three if X is filled by trivalent cations, as required for charge balance (Nagashima et al. 2010). All of these hydroxyl sites involve additional hydrogen bonds, many of which are also bifurcated.
A range of elemental substitutions is possible in pumpellyite. The most common are the exchange of divalent Fe2+ and Mg, and the exchange of trivalent Fe3+ and Al, forming a solid solution series between the idealised end-members pumpellyite-(Mg) [Ca4MgAl5Si6O21(OH)7], and julgoldite-(Fe2+) [Ca4Fe2+Fe3+5Si6O21(OH)7]. Octahedral Y sites contain exclusively trivalent cations, whereas the larger X site contains both divalent and trivalent cations.
Crystal structure of pumpellyite, looking parallel to [010]. Pumpellyite contains four distinct hydroxyl groups with a complex set of hydrogen bonds (dashed lines). If the X site octahedra are solely occupied by trivalent cations, the hydrogen atoms at H5 and H10 are replaced by one at H5′ (Alistair et al. 2017)
Due to the difficulty in distinguishing pumpellyite group members, most specimens are labelled simply as “pumpellyite“, without the chemical suffix. However, sometimes a locality is known to produce one type of member, and this can determine the specific form of pumpellyite.
Pumpellyite was named in 1925 in honor of geologist Raphael Pumpelly (1837-1923), a professor of Mining Science at Harvard University. Pumpelly surveyed the copper region of Michigan, where this mineral was first described.
Acicular olive green pumpellyite-(Fe2+) Ca2Fe”Al2(Si2O7)(SiO4)(OH)2·H2O and green crystals of clinozoisite-epidote {Ca2}{Al3}(Si2O7)(SiO4)O(OH) to {Ca2}{Al2Fe”‘}(Si2O7)(SiO4)O(OH) in vug of altered igneous rock; Los Arenales quarry, Mt. La Rocha de Piquer, Torás, Castellón, Valencian Community, Spain; FOV: 20 mm
Quartz SiO2 crystals covered with white feldspar – albite Na(AlSi3O8) and clusters of pale green pumpellyite-(Mg) Ca2MgAl2(Si2O7)(SiO4)(OH)2·H2O; Helena shaft – Obří důl, (Riesental), Krkonoše Mtn (Riesengebirge), Hradec Králové Region, Czech Republic; 63 × 58 × 30 mm
Remarkable richly mineralized piece of light brown acicular crystals of pumpellyite-(Mn2+) Ca2Mn”Al2(Si2O7)(SiO4)(OH)2·H2O forming veins in association with white quartz SiO2 in beige jasper matrix; Cassagna Mine, Graveglia Valley, Ne, Genova Province, Liguria, Italy; 57 × 41 × 40 mm
Massive altered igneous rock (diorite?) with vugs filled with acicular grey-green pumpellyite-(Fe2+) Ca2Fe”Al2(Si2O7)(SiO4)(OH)2·H2O and green clinozoisite-epidote {Ca2}{Al3}(Si2O7)(SiO4)O(OH) to {Ca2}{Al2Fe”‘}(Si2O7)(SiO4)O(OH); Los Arenales quarry, Mt. La Rocha de Piquer, Torás, Castellón, Valencian Community, Spain; 35 × 57 × 38 mm
Clusters of pale yellow acicular pumpellyite-(Mn2+) Ca2Mn”Al2(Si2O7)(SiO4)(OH)2·H2O crystals on matrix; Valgraveglia Mine, (Gambatesa Mine), Monte Copello, Reppia, Ne, Graveglia Valley, Genova Province, Liguria, Italy; 31 x 26 x 22 mm
Pale-green clusters of acicular pumpellyite-(Mg) Ca2MgAl2(Si2O7)(SiO4)(OH)2·H2O crystals and white albite Na(AlSi3O8) on translucent, colourless quartz SiO2; Helena shaft – Obří důl, (Riesental), Krkonoše Mtn (Riesengebirge), Hradec Králové Region, Czech Republic; FOV: 10 mm
Pale yellow acicular pumpellyite-(Mn2+) Ca2Mn”Al2(Si2O7)(SiO4)(OH)2·H2O crystals on matrix; Valgraveglia Mine, (Gambatesa Mine), Monte Copello, Reppia, Ne, Graveglia Valley, Genova Province, Liguria, Italy; FOV: 20 mm
Light brown acicular crystals of pumpellyite-(Mn2+) Ca2Mn”Al2(Si2O7)(SiO4)(OH)2·H2O in associacion with quartz SiO2 in beige jasper matrix; Cassagna Mine, Graveglia Valley, Ne, Genova Province, Liguria, Italy; FOV: 25 mm
Greyish-white rosettes of pumpellyite-(Al) Ca2Al3(Si2O7)(SiO4)(OH,O)2·H2O from type locality; La Flèche quarry, Bertrix, Luxembourg Province, Belgium; FOV: 25 mm
Off-white sprays up to 1 mm large of pumpellyite-(Al) Ca2Al3(Si2O7)(SiO4)(OH,O)2·H2O on matrix from type locality; La Flèche quarry, Bertrix, Luxembourg Province, Belgium; 40 x 30 x 10 mm
Grey-green pumpellyite-(Fe2+) Ca2Fe”Al2(Si2O7)(SiO4)(OH)2·H2O and grass-green crystals of clinozoisite-epidote {Ca2}{Al3}(Si2O7)(SiO4)O(OH) to {Ca2}{Al2Fe”‘}(Si2O7)(SiO4)O(OH) in vug of altered igneous rock; Los Arenales quarry, Mt. La Rocha de Piquer, Torás, Castellón, Valencian Community, Spain; FOV: 20 mm
Radial spray of off-white acicular crystals of pumpellyite-(Al) Ca2Al3(Si2O7)(SiO4)(OH,O)2·H2O on dark green matrix from the type locality; La Flèche quarry, Bertrix, Luxembourg Province, Belgium; FOV: 20 mm
Veins of light brown acicular pumpellyite-(Mn2+) Ca2Mn”Al2(Si2O7)(SiO4)(OH)2·H2O and white quartz SiO2 in beige jasper matrix; Cassagna Mine, Graveglia Valley, Ne, Genova Province, Liguria, Italy; FOV: 35 mm
Green, minute pumpellyite-(Fe2+) Ca2Fe”Al2(Si2O7)(SiO4)(OH)2·H2O crystals on the surface of white quartz SiO2 with minor reddish hematite Fe2O3; Baveno, Verbano-Cusio-Ossola Province, Piedmont, Italy; 22 x 15 x 14 mm
Acicular green pumpellyite-(Fe2+) Ca2Fe”Al2(Si2O7)(SiO4)(OH)2·H2O on white quartz SiO2 with hematite Fe2O3 crust; Baveno, Verbano-Cusio-Ossola Province, Piedmont, Italy; FOV: 15 mm
Green tufts of pumpellyite-(Fe) Ca2(Fe,Mg)(Al,Fe)2(Si2O7)(SiO4)(OH,O)2·H2O in vacuole of quartz in basaltic matrix; Tichka Massif, High Atlas Mtn, Morocco;
Radial spray of pearlescent acicular crystals of pumpellyite-(Al) Ca2Al3(Si2O7)(SiO4)(OH,O)2·H2O on dark green matrix from the type locality; La Flèche quarry, Bertrix, Luxembourg Province, Belgium; FOV: 20 mm
Julgoldite were named in 1971 by Moore in honor of Julian Royce Goldsmith (26 February 1918, Chicago, Illinois, USA – 23 January 1999, USA), geochemist at the University of Chicago. He was an expert on feldspars and carbonates. He also served as president of the Geochemical Society, the Mineralogical Soeciety of America, and the Geological Society of America.
Dark green-black julgoldite-(Fe2+) Ca2Fe”Fe”’2(Si2O7)(SiO4)(OH)2·H2O and pale green, botryoidal prehnite Ca2Al2Si3O10(OH)2 with massive etched calcite CaCO3; Andesite quarry – Kreimbach-Kaulbach, Wolfstein, Palatinate, Rhineland-Palatinate, Germany; 44 × 28 × 17 mm
Group of acicular julgoldite-(Fe3+) Ca2Fe”’Fe”’2(Si2O7)(SiO4)O(OH)·H2O crystals on platy crystal of stilbite-Ca NaCa4[Al9Si27O72]·nH2O and chalcedony SiO2 matrix; Jalgaon Quarries, Jalgaon District, Maharashtra, India; 42 × 35 × 21 mm
Spectacular and very aesthetic group of julgoldite-(Fe3+) Ca2Fe”’Fe”’2(Si2O7)(SiO4)O(OH)·H2O crystals on platy crystal of stilbite-Ca NaCa4[Al9Si27O72]·nH2O and chalcedony SiO2 matrix; Jalgaon Quarries, Jalgaon District, Maharashtra, India; 42 × 35 × 21 mm
Pale green prehnite Ca2Al2Si3O10(OH)2 spheres on dark green, almost black julgoldite-(Fe2+) Ca2Fe”Fe”’2(Si2O7)(SiO4)(OH)2·H2O and white calcite CaCO3; Andesite quarry – Kreimbach-Kaulbach, Wolfstein, Palatinate, Rhineland-Palatinate, Germany; FOV: 20 mm
Green acicular crystals of julgoldite-(Fe3+) Ca2Fe”’Fe”’2(Si2O7)(SiO4)O(OH)·H2O covered with translucent, colourless chalcedony SiO2; Jalgaon Quarries, Jalgaon District, Maharashtra, India; FOV: 25 mm
Pale green, botryoidal prehnite Ca2Al2Si3O10(OH)2 on dark green, almost black julgoldite-(Fe2+) Ca2Fe”Fe”’2(Si2O7)(SiO4)(OH)2·H2O and white calcite CaCO3; Andesite quarry – Kreimbach-Kaulbach, Wolfstein, Palatinate, Rhineland-Palatinate, Germany; 44 × 28 × 17 mm
Dark Green acicular julgoldite-(Fe3+) Ca2Fe”’Fe”’2(Si2O7)(SiO4)O(OH)·H2O crystals inside and on the surface of platy crystal of stilbite-Ca NaCa4[Al9Si27O72]·nH2O; Jalgaon Quarries, Jalgaon District, Maharashtra, India; stilbite crystal: 33 mm
Microcrystalline dark julgoldite-(Fe2+) Ca2Fe”Fe”’2(Si2O7)(SiO4)(OH)2·H2O on etched calcite CaCO3; Andesite quarry – Kreimbach-Kaulbach, Wolfstein, Palatinate, Rhineland-Palatinate, Germany; FOV: 20 mm
Crusts of dark green julgoldite-(Fe2+) Ca2Fe”Fe”’2(Si2O7)(SiO4)(OH)2·H2O on etched calcite CaCO3; Andesite quarry – Kreimbach-Kaulbach, Wolfstein, Palatinate, Rhineland-Palatinate, Germany; 44 × 28 × 17 mm
Crusts of dark green julgoldite-(Fe2+) Ca2Fe”Fe”’2(Si2O7)(SiO4)(OH)2·H2O revealing primary shape of etched calcite CaCO3; Andesite quarry – Kreimbach-Kaulbach, Wolfstein, Palatinate, Rhineland-Palatinate, Germany; FOV: 25 mm
Shuiskite – mineral of pumpellyite group with chromium on Y possition was named in honor of Vadim Prokopevich Shuisky (Вадима Прокопьевича Шуйского) (b. 1936), a lithologist and researcher of the sedimentary strata of the Urals, Institute of Geology and Geochemistry, Ural Branch of the Russian Academy of Sciences, Ekaterinburg, Russia.
Fiber-like crystals of lilac-grey shuiskite Ca2MgCr2(Si2O7)(SiO4)(OH)2·2H2O with clusters of minute emerald-green uvarovite Ca3Cr2(SiO4)3; Saranovskii Mine, (Saranovskoe), Saranovskaya Village (Sarany), Gornozavodskii area, Permskaya Oblast’, Middle Urals, Urals Region, Russia; FOV: 20 mm
Green, lustrous crystals of uvarovite Ca3Cr2(SiO4)3 on purple shuiskite Ca2MgCr2(Si2O7)(SiO4)(OH)2·2H2O and black chromite Fe”Cr”‘2O4 matrix; Saranovskii Mine, (Saranovskoe), Saranovskaya Village (Sarany), Gornozavodskii area, Permskaya Oblast’, Middle Urals, Urals Region, Russia; 78 × 29 × 16 mm
Well developed crystals of uvarovite Ca3Cr2(SiO4)3 from type locality on lilac-grey sprays of shuiskite Ca2MgCr2(Si2O7)(SiO4)(OH)2·2H2O coating chromitic Fe”Cr”‘2O4 matrix; Saranovskii Mine, (Saranovskoe), Saranovskaya Village (Sarany), Gornozavodskii area, Permskaya Oblast’, Middle Urals, Urals Region, Russia; FOV: 20 mm
Elongated lilac-grey sprays of shuiskite Ca2MgCr2(Si2O7)(SiO4)(OH)2·2H2O with emerald-green uvarovite Ca3Cr2(SiO4)3 garnet on black chromite Fe”Cr”‘2O4 matrix; Saranovskii Mine, (Saranovskoe), Saranovskaya Village (Sarany), Gornozavodskii area, Permskaya Oblast’, Middle Urals, Urals Region, Russia; 34 x 32 x 15 mm
Lazulite (Mg,Fe2+)Al2(PO4)2(OH)2 – named in 1795 by Marten H. Klaproth from the term “Lazaward”, which means heaven in Arabic, in allusion to its colour. Lazulite is in a solid solution series with the mineral scorzalite. The lazulite-scorzalite series ranges from the magnesium rich lazulite to the iron rich scorzalite. The rarer scorzalite does not differ appreciably, except that it tends to be darker, less transparent and denser than lazulite.
Lazulite forms during high grade metamorphism of high silica quartz rich rocks, and in pegmatites. It occurs in association with quartz, andalusite, rutile, kyanite, corundum, muscovite, pyrophyllite and dumortierite, in metamorphic terrains, and with albite, quartz, muscovite, tourmaline and beryl in pegmatites.
Freßnitzgraben in Austria is the type locality of lazulite, with the noteworthy localities of Fischbach, in Styria. In Italy, lazulite comes from the Vizze pass, Bolzano Province; and from Monte Folgorito, Pietrasanta, Lucca Province. Some famous localities in the U.S. are Graves Mountain, Georgia and the Champion Mine in Mono County, California. The locality of Rapid Creek (and nearby Crosscut Creek), in the Yukon Territory of Canada is also well-known for its outstanding transparent dark blue lazulite crystals.
Doubly terminated quartz SiO2 crystals on the dark blue, translucent lazulite (Mg,Fe)Al2(PO4)2(OH)2 and brownish siderite FeCO3; Big Fish River, Dawson mining district, Yukon, Canada; Bigger quartz crystal: 9 mm
Well developed crystals of dark blue lazulite (Mg,Fe)Al2(PO4)2(OH)2 with brown siderite FeCO3 and white to colourless quartz SiO2; Big Fish River, Dawson mining district, Yukon, Canada; biggest crystal: 12 x 12 x 10 mm
Group of deep blue, interpenetrated crystals of lazulite (Mg,Fe)Al2(PO4)2(OH)2 intergrown with brown breunnerite (Mg,Fe)CO3, a variety of ferroan magnesite; Höllgraben, (Imlau), Pfarrwerfen, Werfen, Salzburg, Austria; 71 × 55 × 47 mm
Bluish lazulite (Mg,Fe)Al2(PO4)2(OH)2 masses in quartzite matrix from type locality; Freßnitzgraben, Krieglach, Fischbacher Alpen, Styria, Austria; 27 × 26 × 12 mm
Two blue doubly terminated lazulite (Mg,Fe)Al2(PO4)2(OH)2 crystals in a sandstone matrix; Graves Mountain, Lincoln Co., Georgia, USA; 18 × 13 × 11 mm
Brown breunnerite (Mg,Fe)CO3 (ferroan magnesite) with blue crystals of lazulite (Mg,Fe)Al2(PO4)2(OH)2; Höllgraben, (Imlau), Pfarrwerfen, Werfen, Salzburg, Austria; 71 × 55 × 47 mm
Group of dark blue lustrous crystals to 0.8 cm of lazulite (Mg,Fe)Al2(PO4)2(OH)2 with minute brownish siderite FeCO3 and lustrous quartz SiO2; Big Fish River, Dawson mining district, Yukon, Canada; 84 × 72 × 45 mm
Doubly terminated, blue lazulite (Mg,Fe)Al2(PO4)2(OH)2 crystals in a quartz-rich matrix; Graves Mountain, Lincoln Co., Georgia, USA; bigger crystal: 8 mm long
Well developed faces of lazulite (Mg,Fe)Al2(PO4)2(OH)2 crystal intergrown with brown a variety of ferroan magnesite – breunnerite (Mg,Fe)CO3, in quartz – breunerrite vein in greenschists of the Werfen series (so-called Werfen schists); Höllgraben, (Imlau), Pfarrwerfen, Werfen, Salzburg, Austria; largest crystal: 12 mm
Lustrous crystals of dark blue lazulite (Mg,Fe)Al2(PO4)2(OH)2 on matrix; Rapid Creek, Dawson Mining District, Yukon, Canada; 35 x 20 x 9 mm
The vivianite group forms a group of monoclinic phosphate and arsenate minerals that have very similar structures. Named after one of their more common members, the group is generally very colorful. All members of this group can be very brilliantly colored. Vivianite is famous for its bluish-green color. Erythrite is famous for its red-purple color, and annabergite for its apple-green color.
A general formula of vivianite group minerals stands as:
A2+3(XO4)2 × 8H2O,
where X = P or As and A2+= Co, Fe, Mg, Mn, Ni or Zn.
Schematic representation of the structure of vivianite viewed along the b axis. Black spheres: H2O; white spheres: O2- (after Mori and Ito, 1950)
Annabergite Ni3(AsO4)2 · 8H2O – named by Henry J. Brooke and William Hallowes Miller in 1852 after one of the co-type localities, Annaberg, Saxony, Germany. It has a wonderful, bright green color. This characteristic color is easily noticeable and was used to spot veins of nickel-bearing ore. Annabergite, or Nickel Bloom as it is called by miners, is often found as a green alteration coating on other nickel minerals.
Fragment of radial cluster of pale-green annabergite Ni3(AsO4)2•8H2O; Monte Arsiccio Mine, Sant’Anna di Stazzema, Stazzema, Apuan Alps, Lucca Province, Tuscany, Italy; FOV: 25 mm
Green clusters of transparent annabergite Ni3(AsO4)2•8H2O crystals forming radiating cluster of thin blades; Km-3 Mine, Lavrion, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; FOV: 15 mm
Radial cluster of pale-green annabergite Ni3(AsO4)2•8H2O on matrix; Monte Arsiccio Mine, Sant’Anna di Stazzema, Stazzema, Apuan Alps, Lucca Province, Tuscany, Italy; cluster about 4 mm
Lustrous, bright green crystals of annabergite Ni3(AsO4)2•8H2O in a vug aesthetically placed in matrix; Km-3 Mine, Lavrion, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; FOV: 20 mm
Bright mint green clusters of lustrous annabergite Ni3(AsO4)2•8H2O crystals scattered through an exposed cavity; Km-3 Mine, Lavrion, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; 31 × 19 × 16 mm
Acicular crystals of pale-green annabergite Ni3(AsO4)2•8H2O in radial clusters on matrix; Monte Arsiccio Mine, Sant’Anna di Stazzema, Stazzema, Apuan Alps, Lucca Province, Tuscany, Italy; 46 × 43 × 10 mm
Erythrite
Erythrite Co3(AsO4)2 · 8H2O, also called Cobalt Bloom, arsenate mineral in the vivianite group, hydrated cobalt arsenate. Erythrite, which is used as a guide to the presence of cobalt-nickel-silver ores because of its crimson or peach-red colour, occurs as radiating crystals, concretions, or earthy masses in the oxidized zone of cobalt and nickel deposits. It forms a complete solid-solution series with annabergite, in which nickel replaces cobalt in the erythrite structure. As the nickel content increases, the colour lightens to white, gray, or pale green.
Deep magenta colored terminated crystals of erythrite Co3(AsO4)2•8H2O on white quartz SiO2; Bou Azzer District, Tazenakht, Ouarzazate Province, Drâa-Tafilalet Region, Morocco; FOV: 25 mm
Aggregate of bright magenta crystals of erythrite Co3(AsO4)2•8H2O, very well defined and with good color, that fill in a cavity in a matrix of quartz SiO2 partially covered with chlorite Mg5Al(AlSi3O10)(OH)8; Bou Azzer District, Tazenakht, Ouarzazate Province, Drâa-Tafilalet Region, Morocco; 64 × 47 × 35 mm
Radial group of dark pink crystals of erythrite Co3(AsO4)2•8H2O; La Encontrada Mine, Molvizar, Granada, Andalusia, Spain; FOV: 35 mm
Minute round clusters of magenta erythite Co3(AsO4)2•8H2O on white quartz SiO2 matrix from type locality; Neustädtel, Schneeberg District, Erzgebirge, Saxony, Germany; 25 x 10 x 12 mm
Nice pink erythrite Co3(AsO4)2•8H2O aggregates and microcrystals on matrix; Wittichen, Schenkenzell, Black Forest, Baden-Württemberg, Germany; FOV: 20 mm
Cluster of bright erythrite Co3(AsO4)2•8H2O crystals, very well defined and with good color, that fill in a cavity in a matrix of quartz SiO2 and chlorite Mg5Al(AlSi3O10)(OH)8; Bou Azzer District, Tazenakht, Ouarzazate Province, Drâa-Tafilalet Region, Morocco; FOV: 25 mm
Typical erythrite “cobalt flowers” Co3(AsO4)2•8H2O beige dolomite; Gratlspitz, Brixlegg – Rattenberg, Brixlegg – Schwaz area, Inn valley, North Tyrol, Tyrol, Austria; 20 x 17 x 8 mm
Round clusters of magenta erythite Co3(AsO4)2•8H2O on white quartz SiO2 from type locality; Neustädtel, Schneeberg District, Erzgebirge, Saxony, Germany; 25 x 10 x 12 mm
Purple sprays of erythrite Co3(AsO4)2•8H2O aesthetically placed in vug; Tazalarht mining area, Taroudant Province, Souss-Massa Region, Morocco; FOV: 20 mm
Aggregate of elongated crystals of erythrite Co3(AsO4)2•8H2O that have a quite different shape than the better known erythrites from Bou Azzer Area; Tazalarht mining area, Taroudant Province, Souss-Massa Region, Morocco; FOV: 30 mm
Spheroidal clusters of pink erythrite Co3(AsO4)2•8H2O on matrix; Wenzel Mine, Frohnbach valley, Oberwolfach, Wolfach, Black Forest, Baden-Württemberg, Germany; FOV: 11 mm
Microcrystalline masses of magenta erythrite Co3(AsO4)2•8H2O on matrix; Wittichen, Schenkenzell, Black Forest, Baden-Württemberg, Germany; 29 × 25 × 23 mm
Minute magenta erythrite Co3(AsO4)2•8H2O crystals and microcrystalline clusters on prismatic, white quartz SiO2 crystals; Tazalarht mining area, Taroudant Province, Souss-Massa Region, Morocco; 145 x 81 x 48 mm
Minute clusters of erythrite Co3(AsO4)2•8H2O on matrix; Wenzel Mine, Frohnbach valley, Oberwolfach, Wolfach, Black Forest, Baden-Württemberg, Germany; 22 x 11 x 7 mm
Specimen of acicular pink erythrite Co3(AsO4)2•8H2O and white aragonite CaCO3 crystals on matrix; Gratlspitz, Brixlegg – Rattenberg, Brixlegg – Schwaz area, Inn valley, North Tyrol, Tyrol, Austria; 20 x 17 x 8 mm
Cluster of magenta elongated crystals of erythrite Co3(AsO4)2•8H2O in a vug; Tazalarht mining area, Taroudant Province, Souss-Massa Region, Morocco; FOV: 25 mm
Erythrite Co3(AsO4)2•8H2O microcrystalline clusters between prismatic white quartz SiO2 crystals; Tazalarht mining area, Taroudant Province, Souss-Massa Region, Morocco; FOV: 35 mm
Magenta rossetes of erythrite Co3(AsO4)2•8H2O with white aragonite CaCO3 on dolostone; Gratlspitz, Brixlegg – Rattenberg, Brixlegg – Schwaz area, Inn valley, North Tyrol, Tyrol, Austria; 20 x 17 x 8 mm
Deep red magenta sharply terminated undamaged crystals of erythrite Co3(AsO4)2•8H2O set aesthetically upon a matrix, second generation of erythrite is present on the edges of bigger crystals Bou Azzer District, Tazenakht, Ouarzazate Province, Drâa-Tafilalet Region, Morocco; FOV: 35 mm
Radial sprays of laminar magenta erythrite Co3(AsO4)2•8H2O covering flat surface of the rock; La Encontrada Mine, Molvizar, Granada, Andalusia, Spain; 68 × 34 × 35 mm
Vivianite
Vivianite Fe2+3(PO4)2 · 8H2O – named by Abraham Gottlob Werner in 1817 after John Henry Vivian (August 9, 1785 – February 10, 1855), an English (Welsh-Cornish) politician, mine owner, and mineralogist living in Truro, Cornwall and discoverer of the mineral.
Usually found as deep blue to deep bluish green prismatic to flattened crystals, most crystals rather small to microscopic, larger ones are rather rare. When fresh the mineral may be colourless, or nearly so, and, once exposed, will oxidize with the Fe2+ converting to Fe3+ with a concurrent darkening to dark blue or blue-green.
Two matching parts of black nodule with minute black – dark blue lustrous acicular crystals of vivanite Fe3(PO4)2•8H2O inside; San Giovanni Valdarno, Valdarno (Val d’Arno), Arezzo Province, Tuscany, Italy; 49 x 47 x 31 mm, and 36 x 28 x 17 mm
Fine cluster of lustrous blades of vivianite Fe3(PO4)2•8H2O with very dark green colour with flashes of blue and purple; Kerchenskoe deposit, Kerchenskyi (Fe)-ore basin, Kerch peninsula (Kertch peninsula), Crimea peninsula, Crimea Oblast’, Ukraine; FOV: 35 mm
Acicular crystals of dark blue almost black vivianite Fe3(PO4)2•8H2O; San Giovanni Valdarno, Valdarno (Val d’Arno), Arezzo Province, Tuscany, Italy; FOV: 30 mm
Microcrystalline masses of grey-blue vivianite Fe3(PO4)2•8H2O on matrix; Hagendorf, Waidhaus, Upper Palatinate, Bavaria, Germany; 29 × 13 × 13 mm
A superb small miniature with a perfect, glassy single crystal of green vivianite Fe3(PO4)2•8H2O of superb luster and clarity; Huanuni mine, Huanuni, Dalence Province, Oruro Department, Bolivia; 28 × 4 × 2 mm
Close-up of concretionary nodule with blue, acicular crystals of vivanite Fe3(PO4)2•8H2O; San Giovanni Valdarno, Valdarno (Val d’Arno), Arezzo Province, Tuscany, Italy; FOV: 30 mm
Vivianite Fe3(PO4)2•8H2O in alteration phase to kertschenite FeFe2(PO4)2(OH)2•6H2O, showing metallic blue flash; Kerchenskoe deposit, Kerchenskyi (Fe)-ore basin, Kerch peninsula (Kertch peninsula), Crimea peninsula, Crimea Oblast’, Ukraine; FOV: 45 mm
Lustrous, gem quality, emerald-green, sword-shaped “textbook” crystals of vivianite Fe3(PO4)2•8H2O with minor brownish siderite FeCO3; Huanuni mine, Huanuni, Dalence Province, Oruro Department, Bolivia; 28 × 4 × 2 mm
Well developed crystals of dark (almost black) vivianite Fe3(PO4)2•8H2O are in good condition and sit on limonite containing fragment of nice grown shell; Kerchenskoe deposit, Kerchenskyi (Fe)-ore basin, Kerch peninsula (Kertch peninsula), Crimea peninsula, Crimea Oblast’, Ukraine; 83 × 63 × 37 mm
Fine greenish-black prismatic crystals of vivianite Fe3(PO4)2•8H2O filling a vug (17 x 12 mm) within a limonitic matrix containing remains of carbonate shells of molluscs; Kerch peninsula, Crimea peninsula, Crimea Oblast’, Ukraine; 40 x 38 x 19 mm
Santabarbaraite – vivianite alteration product
Santabarbaraite is relatively new amorphous ferric iron hydroxy phosphate hydrate described from Valdarno, Tuscany, Italy. A simplified chemical formula for the type material can be given as Fe3+3(PO4)2(OH)3·5H2O.
The mineral is the result of in situ oxidation of vivianite, occurring as pseudomorphs after vivianite crystals. Santabarbaraite is brown to light-brown in hand specimen, but appears yellowish amber under the microscope and has a similar streak. It is translucent with a distinct vitreous to greasy luster. It is brittle with a distinct parting along the perfect cleavage of vivianite.
Analysis of santabarbaraite showed that all the Fe in santabarbaraite is trivalent, associated with the presence of both H2O and hydroxyl. This is consistent with an oxidation series from vivianite through metavivianite to santabarbaraite, involving progressive oxidation of Fe2+ accompanied by conversion of H2O ligands to OH ions. Such a process leads to a gradual collapse of the vivianite structure as hydrogen bonds are eliminated. Santabarbaraite is the end product of this process and can be thus considered the phosphorus analogue of ferrisymplesiteFe3+3(AsO4)2(OH)3 · 5H2O (Pratesi et al. 2003).
Fans of red crystals, lanceolate, of santabarbaraite Fe3(PO4)2(OH)3•5H2O pseudomorph after vivianite Fe3(PO4)2•8H2O; Kerchenskoe deposit, Kerchenskyi (Fe)-ore basin, Kerch peninsula, Crimea peninsula, Crimea Oblast’, Ukraine; FOV: 25 mm
Three parts of santabarbaraite Fe3(PO4)2(OH)3•5H2O pseudomorphous after vivianite Fe3(PO4)2•8H2O complete nodule from type locality; Santa Barbara lignite District, Cavriglia, Valdarno, Arezzo Province, Tuscany, Italy; biggest part: 42 × 36 × 34 mm
Santabarbaraite Fe3(PO4)2(OH)3•5H2O pseudomorphs after tabular vivianite Fe3(PO4)2•8H2O crystals in mollusc shell; Kerchenskoe deposit, Kerchenskyi (Fe)-ore basin, Kerch peninsula, Crimea peninsula, Crimea Oblast’, Ukraine; FOV: 25 mm
Santabarbaraite Fe3(PO4)2(OH)3•5H2O pseudomorphous after vivianite Fe3(PO4)2•8H2O from type locality; Santa Barbara lignite District, Cavriglia, Valdarno, Arezzo Province, Tuscany, Italy; 22 × 19 × 14 mm
Brown crystals of santabarbareite Fe3+3(PO4)2(OH)3 · 5H2O inside the shell of calm; Kerch peninsula (Kertch peninsula), Crimea peninsula, Crimea Oblast’, Ukraine; 42 x 40 x 26 mm
Concretionary nodule showing internal cavities filled by aggregates of sub-millimetric pseudocrystals of santabarbaraite Fe3(PO4)2(OH)3•5H2O with the morphology of primary vivianite Fe3(PO4)2•8H2O from type locality; Santa Barbara lignite District, Cavriglia, Valdarno, Arezzo Province, Tuscany, Italy; 42 × 36 × 34 mm
Melilites – a group of tetragonal sorosilicates with a disilicate anion (Si2O7)6- or an Al or B-bearing derivative thereof and the general formula:
Ca2M(XSiO7)
M = Mg, Al, rarely Fe, B, Zn, Be, Si, etc.
X = Si, Al, rarely Be or B
where M denotes a small- to medium-sized divalent or trivalent cation (mostly Mg and Al, or rarely Fe, B, Zn, Be, Si, etc) and X is Si, Al or rarely Be or B. In general, Al or B replace one Si atom when M is a trivalent ion, but the charge can also be balanced by coupled substitution of Ca2+ with a monovalent ion, especially Na, and M3+ with M2+, such as in Alumoåkermanite, where Al3+ is still dominant on the M-site but the mineral is far from end-member composition (mindat.org).
A view down onto the sheets of the Ca2M(XSi2O7) melilite structure. The Ca atoms are yellow spheres, the M sites are pale orange tetrahedra, and the XSi sites are blue tetrahedra. Oxygen atoms (not shown) are on the corners of the tetrahedra; wikipedia.org
In petrology “melilite” usually refers to minerals in the åkermanite–gehlenite series, by far the most abundant members of the group. Melilite with compositions dominated by the endmembers akermanite and gehlenite is widely distributed but uncommon. It occurs in metamorphic and igneous rocks and in meteorites.
Typical metamorphic occurrences are in high-temperature metamorphosed impure limestones. For instance, melilite occurs in some high-temperature skarns.
Melilite also occurs in unusual silica-undersaturated igneous rocks. Some of these rocks appear to have formed by reaction of magmas with limestone. Other igneous rocks containing melilite crystallize from magma derived from the Earth’s mantle and apparently uncontaminated by the Earth’s crust. The presence of melilite is an essential constituent in some rare igneous rocks, such as olivine melilitite – extremely rare igneous rocks contain as much as 70% melilite, together with minerals such as pyroxene and perovskite (wikipedia.org).
Åkermanite Ca2Mg(Si2O7) was first described from samples of slags from furnace iron production found at 3 furnace localities: Hofors, Löfsjöen and Mölnbo, Sweden (Vogt 1884).
Named by the Norwegian geologist, professor Johan Herman Lie Vogt (1858-1932) in honor of Anders Richard Åkerman (1837-1922), Swedish metallurgist. During his comprehensive study of the mineralogy of slag products, Vogt discovered a new Ca-Mg-silicate, and named the mineral Åkermanit. Åkerman had kindly given J.H.L.Vogt access to Stockholms Bergskolas large collection of slags. It was in samples from this collection Vogt discovered the mineral.
GehleniteCa2Al(AlSiO7) was named in 1815 by Johann Nepomuk von Fuchs in honor of Adolf Ferdinand Gehlen [5 September 1775 Bütow, Outer Pomerania, Prussia (Bytów, Poland) – 16 July 1815]. Gehlen was editor and publisher of publisher of Neues Allgemeines Journal der Chemie (1803–06), Journal für Chemie und Physik (1806-10) and the Repetitorium für die Pharmacie. He was initially a chemistry professor at the University of Halle (Martin-Luther-Universität Halle-Wittenberg) and later chemist at the Bavarian Academy of Sciences. He died early due to arsenic poisoning. Type locality for gehlenite are Monzoni Mts in Italy.
White tabular crystals of gehlenite Ca2Al(AlSiO7) in matrix from type locality; Monzoni Mts, Fassa Valley, Trento Province, Trentino-Alto Adige, Italy; FOV: 40 mm
Hexagonal white nepheline (Na,K)AlSiO4 and yellowish translucent leucite K(AlSi2O6) with reddish plates of melilite of åkermanite-gehlenite series and black pyroxene (probably augite); Osa quarry, Osteria dell’Osa, Rome, Rome Province, Latium, Italy; 27 × 29 × 18 mm
Olive-green platy translucent crystal of åkermanite Ca2Mg(Si2O7) in matrix; Vispi Quarry, San Venanzo, Terni Province, Umbria, Italy; crystal about 3 mm
Platy crystals of greenish åkermanite Ca2Mg(Si2O7) in Venanzite; Vispi Quarry, San Venanzo, Terni Province, Umbria, Italy; 24 × 20 × 15 mm
White gehlenite forms a unusually large pseudocubic crystals; Vaţa de Sus, Hunedoara Co., Romania; 60 × 60 × 35 mm
Platy crystal of åkermanite Ca2Mg(Si2O7) in matrix; Vispi Quarry, San Venanzo, Terni Province, Umbria, Italy; 25 × 15 × 10 mm
Platy crystals of åkermanite Ca2Mg(Si2O7) with needle-like kalsilite KAlSiO4 in Venanzite (kalsilite-phlogopite-olivine-leucite melilitite); Vispi Quarry, San Venanzo, Terni Province, Umbria, Italy; 24 × 20 × 15 mm
White platy gehlenite Ca2Al(AlSiO7) crystals; Vaţa de Sus, Hunedoara Co., Romania; FOV: 35 mm
Reddish crystals of melilite of åkermanite-gehlenite series, colourless needle-like fluorapatite Ca5(PO4)3F, white prismatic nepheline (Na,K)AlSiO4 and black amphibole “barkevikite” on volcanic rock; Laghetto quarry, Laghetto, Monte Compatri, Rome Province, Latium, Italy; FOV: 20 mm
Reddish plates of melilite of åkermanite-gehlenite series with yellowish isometric leucite K(AlSi2O6) , white hexagonal nepheline (Na,K)AlSiO4 and black pyroxene on the surface of basaltic leucitite; Osa quarry, Osteria dell’Osa, Rome, Rome Province, Latium, Italy; FOV: 25 mm
Cluster of white gehlenite Ca2Al(AlSiO7) crystals, biggest crystal (on the front of photo) about 15 x 15 x 6 mm; Vaţa de Sus, Hunedoara Co., Romania; FOV: 35 mm
Red crystals of melilite of åkermanite-gehlenite series, colourless needle-like fluorapatite Ca5(PO4)3F, white prismatic nepheline (Na,K)AlSiO4 on matrix; Laghetto quarry, Laghetto, Monte Compatri, Rome Province, Latium, Italy; FOV: 20 mm
Åkermanite-gehlenite series red melilite on matrix; Laghetto quarry, Laghetto, Monte Compatri, Rome Province, Latium, Italy; 40 × 31 × 19 mm
Platy crystal of åkermanite Ca2Mg(Si2O7) with needle-like kalsilite KAlSiO4 in vug of melilitite; Vispi Quarry, San Venanzo, Terni Province, Umbria, Italy; crystal about 3 mm
Tabular olive-green crystals of åkermanite Ca2Mg(Si2O7) in kalsilite – melilitite lava; Vispi Quarry, San Venanzo, Terni Province, Umbria, Italy; 25 × 15 × 10 mm
White tabular crystals of gehlenite Ca2Al(AlSiO7) with largest crystal up to 11 mm from type locality; Monzoni Mts, Fassa Valley, Trento Province, Trentino-Alto Adige, Italy; 47 × 104 × 50 mm
The numerous possible combinations, depending on the elements present at D-, G– and T-sites, led to classify minerals in different groups. The classification proposed by Mills et al. (2009) and refined by Bayliss et al. (2010) consists in a subdivision of the alunite supergroup into various four groups: the alunite group, the plumbogummite group, the dusserite group and the beudantite group.
This division is based on the element that predominates in T-site. Thus, minerals in the alunite group are those for which TO4 is dominated by SO4. Cations in D-site are usually monovalent ions and compounds, with Al3+ as predominant element in G-site, are referred to alunite family, and those with Fe3+ to jarosite family. In the beudantite group, one SO42- is replaced by PO43- or AsO43-, in the plumbogummite group, TO4 is predominated by PO4 and in the dusserite group by AsO4. For these groups, as PO43- or AsO43- increases at the expense of SO42-, charge is compensated by addition of a trivalent cation such as Ce3+, or by protonation of some of the hydroxyl groups in the structure (Maubec et al. 2012).
Alunite family structure exemplified by the atomic arrangement DG3(TO4)2(OH)6in a projection along the c axis. Remarks: TO4 tetrahedra are grey, GO6 octahedra are dark grey, D atoms are black (after Sato et al. 2009).
Crystals in the group are generally small, imperfect, and rare. Habit is usually either tabular {0001} or pseudo-cubic (cube-like rhombohedron) to pseudo-cuboctahedral, or acute-rhombohedral. Acicular habits are extremely rare.
Alunites are a group of minerals, which form part of the alunite supergroup. In the general formula [DG3(TO4)2(OH,H2O)6] the D sites are occupied by monovalent cations such as K, Na, NH4, H3O+ and others, divalent cations such as Ca, Ba, Sr, Pb, trivalent cations for example Bi; and G is the trivalent cation either Al of Fe3+; and T is S6+. Alunites can be divided into alunites and jarosites simply depending on whether the concentration of Al is > Fe (alunites) or Fe > Al (jarosites). Of course, solid solution formation can exist across a wide range of concentrations and substitutions (Frost et al. 2006).
Chunky fragment of alunite matrix with vugs filled with well crystallized reddish-brown lamellar alunite KAl3[(OH)6|(SO4)2]; Montioni, Suvereto, Livorno Province, Tuscany, Italy; 43 × 66 × 43 mm
Alunite KAl3[(OH)6|(SO4)2] crystals on matrix from type locality; Allumiere Quarries, Allumiere, Tolfa Mts District, Rome Province, Latium, Italy; 40 × 23 × 20 mm
Minute reddish lustrous lamellar crystals of alunite KAl3[(OH)6|(SO4)2] on matrix, Montioni, Suvereto, Livorno Province, Tuscany, Italy; 43 × 66 × 43 mm
White to brownish alunite KAl3[(OH)6|(SO4)2] crystals on flat matrix; Bomboly Hill, Mád, Zemplén Mts (Tokaj Mts), Borsod-Abaúj-Zemplén Co., Hungary; 27 × 15 × 12 mm
Crystal cluster of white to brownish (with inclusions of hematite Fe2O3), highly lustrous crystals of alunite KAl3[(OH)6|(SO4)2] to 5 mm; Bomboly Hill, Mád, Zemplén Mts (Tokaj Mts), Borsod-Abaúj-Zemplén Co., Hungary; 27 × 15 × 12 mm
Orange-yellow alunite KAl3[(OH)6|(SO4)2] from type locality; Allumiere Quarries, Allumiere, Tolfa Mts District, Rome Province, Latium, Italy; 40 × 23 × 20 mm
Orange-yellow, coloured with iron oxides alunite KAl3[(OH)6|(SO4)2] from type locality; Allumiere Quarries, Allumiere, Tolfa Mts District, Rome Province, Latium, Italy; 40 × 23 × 20 mm
Sample covered by a layer of white to orange, prismatic alunite KAl3[(OH)6|(SO4)2] crystals from type locality; Allumiere Quarries, Allumiere, Tolfa Mts District, Rome Province, Latium, Italy; 40 × 23 × 20 mm
Reddish lamellar crystals of alunite KAl3[(OH)6|(SO4)2] inside of the vugs, Montioni, Suvereto, Livorno Province, Tuscany, Italy; FOV: 30 mm
White to brownish alunite KAl3[(OH)6|(SO4)2] cluster; Bomboly Hill, Mád, Zemplén Mts (Tokaj Mts), Borsod-Abaúj-Zemplén Co., Hungary; 27 × 15 × 12 mm
Brownish jarosite KFe3[(OH)6|(SO4)2] ‘rosettes’ on goethite FeO(OH); Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; FOV: 15 mm
Interesting combo specimen of light yellowish-brown crystals of natrojarosite NaFe3[(OH)6|(SO4)2] with brown crystals of jarosite KFe3[(OH)6|(SO4)2] on matrix; Christiana Mine, Kamariza Mines (Kamareza Mines), Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; 53 × 45 × 36 mm
Cavity containing off-white masses of kamarizaite Fe3[(OH)3|(AsO4)2]*3H2O rimmed with brown jarosite KFe3[(OH)6|(SO4)2]; Kamariza Mines, Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; FOV: 30 mm
Rich specimen of kamarizaite Fe3[(OH)3|(AsO4)2]*3H2O present all around the specimen as off-white masses with small cavities with brown jarosite KFe3[(OH)6|(SO4)2]; Kamariza Mines, Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; 42 × 40 × 33 mm
Light brown composite jarosite KFe3[(OH)6|(SO4)2] crystals on goethite FeO(OH); Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal;
Brown jarosite KFe3[(OH)6|(SO4)2] crystals richly scattered over goethitic matrix; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; 48 × 33 × 17 mm
Clusters of yellowish microcrystals of natrojarosite NaFe3[(OH)6|(SO4)2] with darker jarosite KFe3[(OH)6|(SO4)2] on matrix; Christiana Mine, Kamariza Mines (Kamareza Mines), Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; 53 × 45 × 36 mm
Several light brown jarosite KFe3[(OH)6|(SO4)2] crystals on matrix; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; 39 × 26 × 21 mm
Cluster of beige, fibrous jarosite KFe3(SO4)2(OH)6; Chuquicamata District, Calama, El Loa Province, Antofagasta Region, Chile; 19 × 8 × 7 mm
Brown jarosite KFe3[(OH)6|(SO4)2] crystals richly scattered over matrix with tiny yellowish corkite PbFe3[(OH)6|SO4|PO4] (beudantite group) crystals on goethite FeO(OH) ; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; FOV: 20 mm
Light yellow plumbojarosite Pb0.5Fe3[(OH)6|(SO4)2] scatered over brown jarosite KFe3[(OH)6|(SO4)2] crystals in cavity; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; FOV: 35 mm
Yellowish-brown microcrystals of natrojarosite NaFe3[(OH)6|(SO4)2] with brown crystals of jarosite KFe3[(OH)6|(SO4)2] on matrix; Christiana Mine, Kamariza Mines (Kamareza Mines), Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; FOV: 20 mm
Superbly crystallized brownish jarosite KFe3[(OH)6|(SO4)2] with multiple twinnings richly scattered over matrix; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; FOV: 20 mm
Sparkling, brown microcrystals of jarosite KFe3[(OH)6|(SO4)2] scattered throughout white sandstone matrix; Mercur District, (Camp Floyd District), Oquirrh Mts, Tooele Co., Utah, USA; 30 × 22 × 19 mm
Triangle vug filled with brown jarosite KFe3[(OH)6|(SO4)2] surrounded with whitish massive kamarizaite Fe3[(OH)3|(AsO4)2]*3H2O from type locality; Kamariza Mines, Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; vug 10 x 5 mm
Very rare, fibrous jarosite KFe3(SO4)2(OH)6; Chuquicamata District, Calama, El Loa Province, Antofagasta Region, Chile; 19 × 8 × 7 mm
Light yellow plumbojarosite Pb0.5Fe3[(OH)6|(SO4)2] scatered over brown jarosite KFe3[(OH)6|(SO4)2] crystals on goethite FeO(OH) matrix; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; 48 × 33 × 17 mm
Turquoise Cu(Al,Fe3+)6(PO4)4(OH)8·4H2O is a secondary mineral occurring in the potassic alteration zone of hydrothermal porphyry copper deposits. May be also formed by the action of meteoric waters, usually in arid regions, on aluminous igneous or sedimentary rocks (as vein filling in volcanic rocks and phosphatic sediments).
and an unnamed Fe2+-Fe3+UM1981-32-PO:FeH analogue Fe2+Fe3+6(PO4)4-x[PO3(OH)]x(OH)8·4H2O (after Foord & Taggart, 1998).
The structure of turquoise group minerals contains distorted MO6 polyhedra (M = Zn, Cu), AlO6 octahedra and PO4 tetrahedra. The dominant structure element is an [Al2MAl2(O,OH,H2O)18] polyhedral cluster formed by a central MO6 polyhedron sharing four edges with two edge-sharing Al(1)O6-Al(2)O6 dimers. Further corner-linkage via the two non-equivalent PO4 tetrahedra forms a three-dimensional frame-work. Hydrogen bonds provide additional links between the polyhedral components (Koltish & Giester, 2000).
View of the turquoise structure along the a-axis (the unit cell is outlined). PO4 tetrahedra are marked with crosses, AlO6 octahedra with parallel lines, and the distorted CuO6 polyhedron is shaded. The large grey sphere represents the partially occupied site (‘X’) at the position (½,0,½). The small grey spheres are H atoms (after Koltish & Giester, 2000)
Turquoise had enormous impact on human civilizations. Joseph E. Pogue (1887-1971) – american geologist, petroleum engineer, and economist, in his book “The Turquoise. A study of its history, mineralogy, geology, ethnology, archaeology, mythology, folkore, and technology” wrote about turquoise: “Turquois is a mineral prized for its perfection of color; for, being opaque, it lacks the brilliant luster that forms the chief attraction of the transparent gems. When of finest quality it possesses a blue tone, soft and pleasing, like the color of clear sky; but often its value is lessened by a greenish cast, and in many stones the green predominates. The mineral is but slightly harder than glass, and may be worked with ease, even by primitive people possessing the crudest tools. (…) At the present day, among civilized nations, the turquois is outranked in value by the diamond, ruby, emerald, sapphire, and some other gems, although over some minds the wonderful blue of that precious stone wields a fascination shared by no other. With many semicivilized peoples, however, the turquois takes foremost rank, and its value depends not only upon its intrinsic worth but also upon the mystic properties and religious signification it is supposed to possess.”
Front page of “The Turquoise. A study of its history, mineralogy, geology, ethnology, archaeology, mythology, folkore, and technology” by Joseph E. Pogue (1915)
Crude turquoise Cu(Al,Fe)6(PO4)4(OH)8·4H2O pseudomorph after fluorapatite crystal; Baviacarr Mine, Municipality of Baviácora, Sonora, Mexico; 35 × 16 × 14 mm
Apple-green wavellite Al3(PO4)2(OH)3·5H2O on bright blue turquoise CuAl6(PO4)4(OH)8·4H2O; Palazuelo de las Cuevas, Aliste, Zamora, Castile and Leon, Spain; 24 × 26 × 17 mm
Cut and polished nodule revealing sky blue segments of turquoise Cu(Al,Fe)6(PO4)4(OH)8 · 4H2O on a brown-white background; Zhilandy, Pavlodar Province, Kazakhstan; 29 × 23 × 10 cm
Turquoise Cu(Al,Fe)6(PO4)4(OH)8·4H2O pseudomorph after fluorapatite crystal; Baviacarr Mine, Municipality of Baviácora, Sonora, Mexico; 35 × 16 × 14 mm
Crusts of cyan turquoise Cu(Al,Fe)6(PO4)4(OH)8·4H2O on the surface of siliceous rock; Palazuelo de las Cuevas, Aliste, Zamora, Castile and Leon, Spain; 79 × 54 × 42 mm
Turquoise Cu(Al,Fe)6(PO4)4(OH)8 · 4H2O, pseudomorph after fluorapatite crystal; Municipality of Baviácora, Sonora, Mexico; 35 x 16 x 11mm
Clusters of sea-green turquoise Cu(Al,Fe)6(PO4)4(OH)8·4H2O on quartzite from classical Polish locality; Pustków Wilczkowski, Kobierzyce, Wrocław District, Strzegom-Sobótka Massif, Lower Silesia (Dolnośląskie), Poland; FOV: 32 x 33 mm
Turquoise Cu(Al,Fe)6(PO4)4(OH)8 · 4H2O; Palazuelo de las Cuevas, Aliste, Zamora, Castile and Leon, Spain; druse about 30 x 12mm
Greenish crusts of botryoidal turquoise Cu(Al,Fe)6(PO4)4(OH)8·4H2O on the surface of quartzite; Pustków Wilczkowski, Kobierzyce, Wrocław District, Strzegom-Sobótka Massif, Lower Silesia (Dolnośląskie), Poland; 97 × 48 × 42 mm
Turquoise Cu(Al,Fe)6(PO4)4(OH)8·4H2O pseudomorph after apatite; Municipality of Baviácora, Sonora, Mexico; 32 mm
Small spheres of teal turquoise Cu(Al,Fe)6(PO4)4(OH)8 · 4H2O on the surface of matrix; Palazuelo de las Cuevas, Aliste, Zamora, Castile and Leon, Spain; globules smaller than 0.5mm
Solid chunk of beautiful blue turquoise Cu(Al,Fe)6(PO4)4(OH)8 · 4H2O with minor matrix; Teghout Cu-Mo deposit, Loṙi Province, Armenia; 28 × 17 × 19 mm
Wavellite Al3(PO4)2(OH,F)3·5H2O (bright green) with turquoise Cu(Al,Fe)6(PO4)4(OH)8·4H2O (teal); Palazuelo de las Cuevas, Aliste, Zamora, Castile and Leon, Spain; druse about 10mm
Rounded massive cluster of blue turquoise Cu(Al,Fe)6(PO4)4(OH)8 · 4H2O with minor matrix; Teghout Cu-Mo deposit, Loṙi Province, Armenia; 28 × 17 × 19 mm
Chrysoberyl Al2BeO4 was discovered in 1789 and described and named by Abraham Gottlob Werner, in 1790. The name ‘chrysoberyl’ comes from the Greek and means ‘gold-colored beryl’. Despite the similarity of their names, chrysoberyl and beryl are two completely different gemstones. Together with alexandrite, chrysoberyl forms an independent gemstone category. Chrysoberyl comes in colors between lemon and greenish yellow, in honey colours, and shades from mint green to brownish green. The colour of yellow chrysoberyl is due to iron substitution. They are mostly found in the gemstone deposits of Brazil, Sri Lanka or East Africa.
In chrysoberyl a common repeated twinning (penetration trilling), especially in the alexandrite variety, causes a stellate appearance and the simulation of hexagonal symmetry. These twinned crystals have a hexagonal appearance, but are the result of a triplet of twins with each “twin” oriented at 120° to its neighbors and taking up 120° of the cyclic trilling.
Examples of typical alexandrite twinning (penetration trilling), Izumrudnye Kopi area, Russia, acc. to “Atlas der Krystallformen” Goldschmidt 1918
If only two of the three possible twin orientations are present, a “V”-shaped twin results. Frequently an inclusion known as a stepped twin can be seen.
Morphology of V-shaped twin of chrysoberyl from Pratinhas, Brazil. (a) The twin boundary follows a (130) contact plane, splitting the crystal on two mirror-symmetric halves, with the two domains enclosing an angle of ∼59.5°. (b) Schematic illustration of (130) twin of chrysoberyl (after Drev et al. 2015)
TEM and electron diffraction study of (130) twin boundary in chrysoberyl from Pratinhas. (a) Bright-field TEM image of the twin boundary intersecting thin crystal part populated with oriented TiO2 precipitates. Electron diffraction pattern (EDP) recorded in domain I is rotated by ∼59.5° with respect to that of domain II, which exactly corresponds to the angle enclosed by the a-axes (marked by arrows), distinctive for (130) twin. (b) Twin boundary step to adjacent (130) planes. Note the moiré pattern in the transition area, produced by overlapping crystals in twin orientation. (c) Simulated EDP for (130) twin of chrysoberyl in [001] orientation based on crystallographic data obtained from Rietveld refinement (after Drev et al. 2105)
The best-known special effect of chrysoberyl is an eye, which is displayed when certain specimens of this gem are cut in a dome shape. Cat’s-eye chrysoberyl has a pupil-like band of light that sweeps across its dome. The “eye” is caused by fibrous inclusions that reflect the light in a sharply defined pattern. Chrysoberyl cat’s eyes are genuine rarities which are found only in a few deposits in the world, together with other varieties of chrysoberyl.
Beautiful free-standing trapiche chrysoberyl BeAl2O4 crystal with several other crystals on quartz SiO2 matrix; Ampanorana Est, Ambatondrazaka District, Alaotra-Mangoro Region, Toamasina Province, Madagascar; 32 × 18 × 23 mm
Twinned crystal of green chrysoberyl BeAl2O4 with distinct striation from old collection of Bob Janssens; Rio das Pratinhas, Arataca, Bahia, Brazil, 22 × 17 × 12 mm
Partially visible, star-shaped repeated twinning (penetration trilling) creating faux hexagonal symmetry of yellow-green chrysoberyl BeAl2O4 in quartz SiO2; Ampanorana Est, Ambatondrazaka District, Alaotra-Mangoro Region, Toamasina Province, Madagascar; 32 × 18 × 23 mm
Green-yellow chrysoberyl crystal with well visible twin in sillimanite pegmatite; Rasovna, Maršíkov, Šumperk, Olomouc Region, Moravia, Czech Republic; 65 × 45 × 30 mm
V-shape twin of chrysoberyl BeAl2O4 with distinct striation and muscovite KAl2(Si3Al)O10(OH)2 crystal from old collection; Rio das Pratinhas, Arataca, Bahia, Brazil, 22 × 17 × 12 mm
Lustrous twinned chrysoberyl BeAl2O4 crystals with muscovite KAl2(Si3Al)O10(OH)2 from classic Brazilian locality acquired in 1977; Rio das Pratinhas, Arataca, Bahia, Brazil, 22 × 17 × 12 mm
Gemmy and lustrous, flattened, yellow-green chrysoberyl BeAl2O4 crystal in sillimanitic pegmatite matrix from classical Czech locality; Rasovna, Maršíkov, Šumperk, Olomouc Region, Moravia, Czech Republic; 65 × 45 × 30 mm
Yellow-green chrysoberyl BeAl2O4 V-shape twinned crystals with pseudohexagonal muscovite KAl2(Si3Al)O10(OH)2 from classic Brazilian locality acquired in 1977; Rio das Pratinhas, Arataca, Bahia, Brazil, 22 × 17 × 12 mm
Historic chrysoberyl specimen with 8 mm big gemmy green-yellow crystal in quartz-sillimanite matrix from classical Czech locality; Rasovna, Maršíkov, Šumperk, Olomouc Region, Moravia, Czech Republic; 65 × 45 × 30 mm
Alexandrite, the chromium-bearing variety, is extremely rare and highly prized as a gemstone because of its unique colouring. It is green by daylight and red under ordinary incandescent illumination; it is also strongly pleochroic.
Dark, almost black, partially twinned chrysoberyl var alexandrite BeAl2O4 crystals with purple tint in artificial (incandescent light bulb) light embedded in schist matrix; Carnaíba mining district, Pindobaçu, Campo Formoso ultramafic complex, Bahia, Brazil; crystal 13 x 7 x 3 mm
Chrysoberyl var alexandrite BeAl2O4 crystal showing fairly nice trilling form and green colour in natural and LED light from a classic locality; Izumrudnye Kopi area, Malyshevo, Ekaterinburg, Sverdlovskaya Oblast’, Middle Urals, Urals Region, Russia; 31 x 17 x 14 mm
Twinned crystals of dark, almost black, chrysoberyl var alexandrite BeAl2O4 crystals embedded in schist matrix with purple tint in artificial (incandescent light bulb) light; Carnaíba mining district, Pindobaçu, Campo Formoso ultramafic complex, Bahia, Brazil; crystal 38 × 25 × 15 mm
Dark, almost black, partially twinned chrysoberyl var alexandrite BeAl2O4 crystals with greenish tint in natural and LED light embedded in schist matrix; Carnaíba mining district, Pindobaçu, Campo Formoso ultramafic complex, Bahia, Brazil; crystal 13 x 7 x 3 mm
Dark, almost black, partially twinned chrysoberyl var alexandrite BeAl2O4 crystals embedded in schist matrix. Visible colour change dependant from light type – greenish tint in natural and LED light, purple tint in artificial (incandescent light bulb) light; Carnaíba mining district, Pindobaçu, Campo Formoso ultramafic complex, Bahia, Brazil; crystal 13 x 7 x 3 mm
Chrysoberyl var alexandrite BeAl2O4 crystal showing fairly nice trilling form and brownish-red colour in artificial (incandescent light bulb) light from a classic locality; Izumrudnye Kopi area, Malyshevo, Ekaterinburg, Sverdlovskaya Oblast’, Middle Urals, Urals Region, Russia; 31 x 17 x 14 mm
Twinned crystals of dark, almost black, chrysoberyl var alexandrite BeAl2O4 crystals embedded in schist matrix with greenish tint in natural and LED light; Carnaíba mining district, Pindobaçu, Campo Formoso ultramafic complex, Bahia, Brazil; 38 × 25 × 15 mm
The term “sulfosalt” (or “thiosalt”) was created by chemists during the XIXth century, by analogy to complex salts of oxygen, such as sulfate, phosphate, arsenate, antimonate, arsenite and antimonite. Oxysalts generally correspond to the combination of a simple cation with a complex anion (MeOm)n−; this has been confirmed by crystal-structure studies and bond-valence calculations. In sulfosalts, S is considered to play the role of oxygen to similarly form complex anions. Sulfosalt minerals form a genetically well-defined group encountered in specific conditions of ore formation, usually referred to as hydrothermal processes (Moëlo et al. 2008).
Table presents the hierarchical structure of the system used by Moëlo et al. 2008. Whenever possible, the system is based on the level of structural relationships among mineral species. Thus, for a large number of species the system is essentially a modular classification.
Classification hierarchy of sulfosalts (Moëlo et al. 2008)
More than 260 mineral species belong to the “sulfosalt group” (sulfosalts and other chalcogeno-salts). There are also about 200 incompletely defined minerals (so-called “UM” – unnamed minerals) in the literature related to this vast group (Smith & Nickel, 2007), mainly because the chemical composition alone was determined by EPMA, which is generally easier to obtain than crystallographic data (Moëlo et al. 2008).
The “sulfosalt group” is as heterogeneous from a crystal-chemical point of view as, e.g., the silicate group. Consequently, a rigorous classification and nomenclature of sulfosalts is much more complicated than that of more restricted mineral groups which have been reexamined in the past by specific committees of the IMA (amphiboles, micas, zeolites…). As the bulk of natural thioarsenites, thiostannates, etc. corresponds structurally to homeotypes of simple sulfides (e.g. arsenopyrite), the term “sulfosalt” is usually limited to the vast group of chalcogeno-salts containing trivalent As, Sb or Bi, as well as (exceptionally) Te4+. The S2− anion may be replaced by Se2− or Te2− (chalcogeno-salts). Thus, the general chemical formula can be given as:
At the present stage of research, some groups of these sulfosalts can already be neatly classified on a crystal-chemical basis, whereas others await further discoveries for achieving the same depth of classification. The latter are grouped on purely chemical principles (Moëlo et al. 2008).
Steel gray, cylindrical crystals of cylindrite Pb3Sn4FeSb2S14 with brownish wurtzite ZnS; Poopó town, Poopó Province, Oruro Department, Bolivia; FOV: 40 mm
Sharp, lustrous, gray freibergite Ag6Cu4Fe2Sb4S13-x crystals nicely placed on a tan siderite FeCO3 crystals; Obecnice, Příbram, Central Bohemia Region, Bohemia, Czech Republic; FOV: 30 mm
Cogwheel shape crystals of gray bournonite PbCuSbS3 with golden chalcopyrite CuFeS2 and bright brassy pyrite FeS2; Baia Sprie mine (Felsöbánya mine), Baia Sprie (Felsöbánya), Maramureș Co., Romania; crystal 11 x 8 x 5 mm
Group of iridescent metallic emplectite CuBiS2 needles in a vugh in quartz SiO2 matrix; Barbora adit, Knöttel area, Krupka, Krušné Hory Mts, Ústí Region, Bohemia, Czech Republic; FOV: 15 mm
Light gray metallic needles of mixture of rare sulphosalts bismuthinite Bi2S3 and gladite PbCuBi5S9 in white quartz SiO2; Vyšná Boca, Liptovský Mikuláš Co., Žilina Region, Slovakia; cluster about 20 mm
Rich aggregate of prismatic dark steel-grey berthierite FeSb2S4 aggregates with deep blue iridescent tarnish in yellow-red matrix; Vlastějovice, Zruč nad Sázavou, Central Bohemia Region, Bohemia, Czech Republic; FOV 55 mm
Needle-like brass color crystals of plumosite (boulangerite Pb5Sb4S11 or jamesonite Pb4FeSb6S14) on dark gray calcite CaCO3; Herja Mine, Chiuzbaia, Baia Mare, Maramureș Co., Romania; FOV: 35 mm
Cluster of dark, lead-grey, lustrous mass of franckeite Pb21.7Sn9.3Fe4.0Sb8.1S56.9; San José Mine, Oruro City, Cercado Province, Oruro Department, Bolivia; 82 x 61 x 36 mm
Several platy, metallic crystals of telluronevskite Bi3TeSe2 on a lightly colored matrix; Poruba pod Vihorlatom, Michalovce Co., Košice Region, Slovakia; biggest crystal about 1 mm
Blue iridescent needles of emplectite CuBiS2 in quartz SiO2 druse; Barbora adit, Knöttel area, Krupka, Krušné Hory Mts, Ústí Region, Bohemia, Czech Republic; FOV: 25 mm
Small aggregates of steel-gray telluronevskite Bi3TeSe2 plates in matrix; Poruba pod Vihorlatom, Michalovce Co., Košice Region, Slovakia; FOV: 15 mm
Rich specimen of metallic-bright, elongated, cylindrite FePb3Sn4Sb2S14 crystals on matrix; Poopó town, Poopó Province, Oruro Department, Bolivia; FOV: 35 mm
Aesthetic specimen of bluish iridescent berthierite FeSb2S4 in red-yellow matrix; Vlastějovice, Zruč nad Sázavou, Central Bohemia Region, Bohemia, Czech Republic; 83 x 61 x 36 mm
Lustrous, metallic, cylindrical crystals of cylindrite Pb3Sn4FeSb2S14; Poopó town, Poopó Province, Oruro Department, Bolivia; 72 x 30 x 25 mm
Black-grey grains of very rare telluronevskite Bi3TeSe2 in bright matrix from type locality; Poruba pod Vihorlatom, Michalovce Co., Košice Region, Slovakia; 36 x 28 x 26 mm
Elongated crystal of gray bournonite PbCuSbS3 with golden chalcopyrite CuFeS2 and bright brassy pyrite FeS2; Baia Sprie mine (Felsöbánya mine), Baia Sprie (Felsöbánya), Maramureș Co., Romania; crystal 5 mm long
Tetrahedric freibergite Ag6Cu4Fe2Sb4S13-x crystals sitting in limonite matrix; Obecnice, Příbram, Central Bohemia Region, Bohemia, Czech Republic; biggest crystal about 3 mm
Rosette of stacked crystals of calcite CaCO3, colored deep gray from the included boulangerite Pb5Sb4S11; Herja Mine, Chiuzbaia, Baia Mare, Maramureș Co., Romania; 7 x 7 x 5 mm
Rich aggregate of berthierite FeSb2S4 with deep blue iridescent tarnish; Vlastějovice, Zruč nad Sázavou, Central Bohemia Region, Bohemia, Czech Republic; 83 x 61 x 36 mm
Small dark red crystals of proustite Ag3AsS3 on the matrix of gray rammelsbergite NiAs2; Měděnec, Klášterec nad Ohří, Krušné Hory Mts, Ústí Region, Bohemia, Czech Republic; 21 x 18 x 9 mm
Quartz var. Marmarosh Diamond (“Herkimer-style” quartz) SiO2 with inclusions of boulangerite (var.plumosite) Pb5Sb4S11; Herja Mine, Chiuzbaia, Baia Mare, Maramureș Co., Romania; 41 x 37 x 20 mm
Red crystals of proustite Ag3AsS3 on gray rammelsbergite NiAs2; Měděnec, Klášterec nad Ohří, Krušné Hory Mts, Ústí Region, Bohemia, Czech Republic; 21 x 18 x 9 mm
Fibrous crystals of brown plumosite (boulangerite Pb5Sb4S11 or jamesonite Pb4FeSb6S14) on calcite CaCO3 colored with inclusions of boulangerite Pb5Sb4S11; Herja Mine, Chiuzbaia, Baia Mare, Maramureș Co., Romania; 73 x 68 x 59 mm
An extremely rare mineral cylindrite FePb3Sn4Sb2S14 with a unique cylindrical habit from the type locality; Poopó town, Poopó Province, Oruro Department, Bolivia; bigest crystal 19 mm long
Sharp, lustrous, gray freibergite Ag6Cu4Fe2Sb4S13-x crystals nicely placed on a tan siderite FeCO3 and dark limonite matrix; Obecnice, Příbram, Central Bohemia Region, Bohemia, Czech Republic; 65 x 53 x 34 mm
Cogwheel shape crystals of gray bournonite PbCuSbS3 with golden chalcopyrite CuFeS2 and bright brassy pyrite FeS2; Baia Sprie mine (Felsöbánya mine), Baia Sprie (Felsöbánya), Maramureș Co., Romania; 51 x 34 x 31 mm
Small dictionary:
The crystal structure of two compounds are isotypicif their atoms are distributed in a like manner and if they have the same symmetry. One of them can be generated from the other one if atoms of an element are substituted by atoms of another element without changing their position in the crystal structure. The absolute values for the lattice dimensions and the interatomic distancesmay differ, and small variations are permitted for the atomic coordinates. The angles between the crystallographic axes and the relative lattice dimensions (axes ratios) must be similar. Two isitypic structures exhibit a one-to-one relation for all atomic positions and have coincident geometric conditions (Müller 1991). Isotypic structure means, that mineral have analogous composition and closely similar crystal structure, but is not capable of intercrystallizing to form solid solutions. Examples are calcite and soda niter; galena and NaBr (mindat.org).
Two structures are homeotypic if they are similar, but fail to fulfil the aforementioned conditions for isotypism because of different symmetry, because corresponding atomic positions are occupied by several different kinds of atoms (substitution derivatives) or because the geometric conditions differ (different axes ratios, angles, or atomic coordinates) (Müller 1991).
A homology is a series of structures built on the same structural principle with certain module(s) expanding in various dimension(s) by regular increments. This could be through the addition of a layer or row of atoms on a given module. If the module assembly principle is constant and only the module size and volume evolve, then we can view the series as a set of isoreticular compounds (Kanatzidis 2004).
Merotypic structures (merotypes; meros = part) are composed of alternating layers (blocks), in the same way as do the homologous series. However, one set of these building layers (blocks) are common to all merotypes (i.e. they are isotypic, homeotypic or they are mutually related via homologous expansion/contraction) whereas layers (blocks) of the other set(s) differ for different mesotypes (Merlino 1997).
Plesiotypic structures (plesiotypes; plesios = near, close) form a group, that is built on the same overall principles. It means that (a) they contain fundamental structural elements (blocks, layers) of the same general type(s) and (b) mutual disposition/interconnection of these elements in all plesiotypes follows the same general rules. However, unlike the homologuous series, (1) the plesiotypic structures may contain additional structural elements that differ from one member of the family to another; (2) details of fundamental elements may may also differ between distinct members of one plesiotypic family; within this family, such elements may be interrelated by means of homologous or non-homologousexpansion, truncation, slip planes across them or in their interior, etc.; (3) details of the relationships sub (b) may differ as well (Merlino 1997).
References:
Moëlo Y., Makovicky E., Mozgova N.N., Jambor J.L., Cook N., Pring A., Paar W., Nickel E.H., Graeser S., Karup-Møller S., Balic-Žunic T., Mumme W.G., Vurro F., Topa D., Bindi L., Bente K. & Shimizu M. (2008): Sulfosalt systematics: a review. Report of the sulfosalt sub-committee of the IMA Commission on Ore Mineralogy. Eur. J. Mineral. 20, 7–46.
Crystal habit of wiluite from Vilyui River Basin, Eastern-Siberian Region, Russia; acc. to “Atlas der Krystallformen” Goldschmidt 1918
The vesuvianite group consists of four varieties, i.e. vesuvianite (Mg, OH-rich), wiluite (B-rich) manganvesuvianite and fluorvesuvianite. This mineral group is characteristic for its extremely complex chemical composition, manifold exchange mechanisms and cation ordering, the latter responsible for deviations from the P4/nnc symmetry and for typical polytypic arrangements (Balassone et al. 2011).
Formula: X19Z13T5(Si2O7)4(SiO4)10A10
X = Ca, Na, REE, Pb2+, Sb3+, ☐ Z = Al, Mg, Fe3+, Fe2+, Ti, Mn3+, Cu, Zn T = B, Al, Fe3+, ☐ A = OH, F, O
Individual members of this group are difficult to distinguish without detailed analyses.
Originally named “hyacinthus dictus octodecahedricus” by Moritz Anton Cappeler in 1723. Renamed “hyacinte du Vesuve” by Jean-Baptiste Louis Romé de L’Isle in 1772. This was possibly the inspiration for Abraham Gottlob Werner to rename the species “vesuvian” in 1795, after its discovery locality, Mount Vesuvius, Campania, Italy. In 1799, Rene Just Haüy introduced the name “idocrase”, which was formerly a popular name (mindat.org).
Vesuvianite is a fairly common rock-forming or accessory Ca, Al, Fe, Mg-bearing silicate found in a wide range of occurrences, like thermally metamorphosed limestones or metasomatic rocks (hornfels, skarns), in regional metamorphosed calc-silicate rocks, in rodingites and metarodingites, in metasomatized silicaundersaturated nepheline syenites. Due to its abyssophobic feature, it is normally not observed in high-pressure environments, like blueschist- or eclogite-facies rocks.
Brown, elongated crystals of vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 with green clinochlore Mg5Al(AlSi3O10)(OH)8 and white calcite CaCO3; Canari Mine, Canari, Bastia, Haute-Corse, Corsica, France; longest crystal 22mm
Lustrous, clear, double terminated reddish brown vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 crystal on black clinochlore Mg5Al(AlSi3O10)(OH)8 crystals; Fee Glacier, Saas-Fee, Saas Valley, Zermatt – Saas Fee area, Wallis, Switzerland; central vesuvianite crystal about 2 mm long
Brown clusters of elongated crystals of vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 with platy green clinochlore Mg5Al(AlSi3O10)(OH)8 and white calcite CaCO3; Canari Mine, Canari, Bastia, Haute-Corse, Corsica, France; FOV 35 mm
Lustrous, partially gemmy, olive-green vesusvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 crystal; Crestmore, Riverside Co., California, USA; 15 x 6 mm
Lustrous red-brown vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 crystal on dark clinochlore Mg5Al(AlSi3O10)(OH)8; Fee Glacier, Saas-Fee, Saas Valley, Zermatt – Saas Fee area, Wallis, Switzerland; crystal about 2 mm
Single crystal of lustrous, partially gemmy, olive-green vesusvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10; Crestmore, Riverside Co., California, USA; 15 x 6 mm
An elegant cluster of deep wine-red vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10; Alchuri, Shigar Valley, Skardu District, Baltistan, Gilgit-Baltistan, Pakistan; longest crystal 10 mm
Sharp, slightly translucent, lustrous, terminated vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 crystal cluster; Alchuri, Shigar Valley, Skardu District, Baltistan, Gilgit-Baltistan, Pakistan; 12 x 11 x 7 mm
Cluster of elongated crystal of vesuvianite (egeran) (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 in matrix; Hazlov, Cheb, Karlovy Vary Region, Bohemia, Czech Republic; FOV 35 mm
A few very lustrous red-brown barrel shaped vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 crystals on a carpet of dark green clinochlore Mg5Al(AlSi3O10)(OH)8 and associated with some elongated white pyroxene var. diopside CaMgSi2O6 crystals; Fee Glacier, Saas-Fee, Saas Valley, Zermatt – Saas Fee area, Wallis, Switzerland; 55 x 53 x 28 mm
Cinnamon-colored garnet var. grossular Ca3Al2(SiO4)3 and vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 on matrix; Hazlov, Cheb, Karlovy Vary Region, Bohemia, Czech Republic; 50 x 37 x 37mm
Brown crystals of vesuvianite (called here egeran)(Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 and orange garnet var. grossular Ca3Al2(SiO4)3; Hazlov, Cheb, Karlovy Vary Region, Bohemia, Czech Republic; 50 x 37 x 37 mm
Dark brown, elongated crystals of vesuvianite (egeran) (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 on matrix; Hazlov, Cheb, Karlovy Vary Region, Bohemia, Czech Republic; 50 x 37 x 37 mm
A very attractive specimen with classic stacked up crystals of vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 of deep chocolate brown color; Alchuri, Shigar Valley, Skardu District, Baltistan, Gilgit-Baltistan, Pakistan; 12 x 11 x 7 mm
Lustrous brown vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10 crystals in matrix of massive blue calcite CaCO3 and yellowish fluorellestadite Ca5(SiO4)1.5(SO4)1.5F; Crestmore quarries, Crestmore, Riverside Co., California, USA
Grossular var. rosolite Ca3Al2(SiO4)3 with beige vesuvianite (Ca,Na)19(Al,Mg,Fe)13(SiO4)10(Si2O7)4(OH,F,O)10; Sierra de Cruces, Mun. de Sierra Mojada, Coahuila, Mexico; FOV: 35 mm
In an investigation of vesuvianite from a wide variety of locations around the world (Groat 1988, Groat et al. 1992), it was confirmed that boron could be a major constituent in vesuvianite. The amounts of boron found are quite large (vesuvianite can contain significant amounts of boron up to ~4 wt. % of B2O3), and have significant effects on the bulk chemistry and thermal stability of the mineral. In addition vesuvianite can be a sink for boron in those environments in which it occurs, and hence may play a role in the boron cycle (Groat et al. 1994).
Vesuvianite from the Vilyui River has long been known to have a high boron content, and the work of Groat et al. (1994) showed that vesuvianite from this locality contains sufficient boron to establish a new species. The new mineral is named wiluite, after the locality, the Vilyui River Basin, Sakha Republic, Eastern-Siberian Region, Russia. The new mineral and mineral name have been approved by the International Mineralogical Association Commission on New Minerals and Mineral Names. Type material is deposited at the Canadian Museum of Nature, Ottawa, Ontario, Canada (Groat et al. 1998).
Sharp, euhedral dark blackish-green color prismatic tetragonal crystals of wiluite Ca19(Al,Mg)13(B,☐,Al)5(Si2O7)4(SiO4)10(O,OH)10 in matrix; Vilyui River Basin, Sakha Republic, Eastern-Siberian Region, Russia; 38 x 30 x 19 mm
Tetragonal, well-terminated, lustrous, blackish-green crystals of wiluite Ca19(Al,Mg)13(B,◻,Al)5(SiO4)10(Si2O7)4(O,OH)10 aesthetically perched on top of its matrix; Vilyui River Basin, Sakha Republic, Eastern-Siberian Region, Russia; biggest crystal 3 x 2 x 2 mm
Sharp, glassy, partially translucent, doubly terminated, mottled olive-green crystal of wiluite Ca19(Al,Mg)13(B,◻,Al)5(SiO4)10(Si2O7)4(O,OH)10aesthetically set in matrix; Vilyui River Basin, Sakha Republic, Eastern-Siberian Region, Russia; FOV 30 mm
References:
Balassone G., Talla D., Beran A., Mormone A., Altomare A., Moliterni A., Mondillo N., Saviano M. & Petti C. (2011): Vesuvianite from Somma-Vesuvius volcano (southern Italy): chemical, X-ray diffraction and single-crystal polarized FTIR investigations. Periodico di Mineralogia 80, 3 (Spec. Issue), 369-384.
Groat L.A., Hawthorne F., Ercit T.S. (1994): The Incorporation Of Boron Into The Vesuvianite Structure. The Canadian Mineralogist 32, 505-523.
Groat L.A., Hawthorne F., Ercit T.S. & Grice J.D. (1998): Wiluite Ca19(Al,Mg,Fe,Ti)13(B,Al,☐)5Si18O68(O,OH)10 A New Mineral Species Isostructural With Vesuvianite From The Sakha Republic, Russian Federation. The Canadian Mineralogist 36, 1301-1304.
REE fluorcarbonates group are also known as the bastnäsite series (Donnay & Donnay, 1953). The group consists of four minerals: bastnäsite (REEFCO3), synchysite (REEFCO3 · CaCO3), parisite (2REEFCO3 · CaCO3), and röntgenite (3REEFCO3 · 2CaCO3).
Bastnäsite accounts for approximately 90% of the world’s REE production; synchysite occurs subordinately and is associated with bastnäsite. In almost all the cases where bastnäsite is exploited, parisite and röntgenite are comparatively rare.
The general mineral formula for the group is nXYCO3 · mCaCO3, where:
X = LREE,
Y = (F, OH)
m = 0 (bastnäsite) or 1 (synchysite, parisite, röntgenite),
n = 1 (bastnäsite, synchysite), 2 (parisite) or 3 (röntgenite)
Synchysite displays monoclinic symmetry, whereas other species of the bastnäsite group show trigonal or hexagonal symmetry.
The bastnäsite minerals group present two common features:
the structure can be broadly described as the stacking of three types of layers (CeF, CO3 and Ca) along the c axis;
the Ce/F ratios in all the phases are 1, indicating that this feature is common in all minerals of the group.
According to Donnay & Donnay (1953), in most cases, fluorcarbonates are polycrystals with syntaxial intergrowth of two species in contact along an irregular surface or along repeated parallel planes (0001). All pairs have been observed, except the bastnäsite-synchysite pair.
Van Landuyt & Amelinckx (1975) claim, that the syntactic intergrowths can be described as mixtures of bastnäsite and synchysite. The authors considered bastnäsite-(Ce) and synchysite-(Ce) as two end-members and that parisite and röntgenite are ordered mixtures of bastnäsite (B) and synchysite (S) in single layers stacked along the c crystallographic axis direction. Parisite can be considered a BS stacking and röntgenite a BS2 stacking (Manfredi et al. 2013).
Atomic arrangement of bastnäsite-(Ce), synchysite-(Ce) and parisite-(Ce) projected on (010). Triangles represent (CO3) groups, and O atoms lie at the apices of the triangles. Circles from the largest to the smallest represent F, Ce, and Ca atoms, respectively. The unit cells are outlined.
Additional REE minerals that commonly occur in fluorocarbonate-bearing REE deposits include monazite {REEPO4}, allanite {(Ca,REE)2(Al,Fe,Mg)3Si3012(OH)}, ancylite {REESr(CO3(OH)·H2O}, burbankite {(REE,Na,Ca,Sr,Ba)6(CO3)5}, calkinsite {REE2(CO3)3·4H2O} , lanthanite {REE2(CO3)3·8H2O}, and fluocerite {REEF3}.
Bastnäsite Group minerals
The name came from the type locality at the Bastnäs mines, Riddarhyttan, Skinnskatteberg, Västmanland, Sweden. The most common member of this group is bastnäsite-(Ce). F-enriched species in this group can form in an environment relatively low in F content, whereas OH-species are rare and occur only in low-temperature environments essentially devoid of F (Hsu, 1992).
Complete hexagonal crystal of bastnäsite-(Ce) CeCO3F with inclusions of black aegirine NaFeSi2O6 and yellowish rutile TiO2; Zagi Mountain, Hameed Abad Kafoor Dheri, Peshawar, Khyber Pakhtunkhwa, Pakistan; crystal: 11 x 8 x 6 mm
Pinkish to orange balls of Bastnäsite-(Ce) CeCO3F; La Flèche quarry, Bertrix, Luxembourg Province, Belgium; FOV 12 mm
Yellowish pseudomorph of bastnäsite-(Y) YCO3F after gagarinite-(Y) NaCaYF6 in quartz core of alkaline granitic pegmatite from type locality; Verkhnee Espe Massif, Akzhaylyautas Mts, Tarbagatai Range, Eastern Kazakhstan Province, Kazakhstan; 25 x 13 x 8 mm
Honey-orange hexagonal crystal of bastnäsite-(Ce) CeCO3F with sharp faces and good luster associated with a white orthoclase var. adularia KAlSi3O8 crystals; Zagi Mountain, Hameed Abad Kafoor Dheri, Peshawar, Khyber Pakhtunkhwa, Pakistan; crystal: 11 x 8 x 6 mm
Pseudomorph of yellowish bastnäsite-(Y) YCO3F after gagarinite-(Y) NaCaYF6 crystals in quartz SiO2 core of alkaline granitic pegmatite from type locality; Verkhnee Espe Massif, Akzhaylyautas Mts, Tarbagatai Range, Eastern Kazakhstan Province, Kazakhstan; 25 x 13 x 8 mm
Bastnäsite-(Ce) Ce(CO3)F in kaolinite Al2(Si2O5)(OH)4 (darkest) and muscovite (var. sericite) KAl2(AlSi3O10)(OH)2; Miłków, Jelenia Góra District, Lower Silesia (Dolnośląskie), Poland; SEM-BSE image
Synchysite Group minerals
Named in 1901 by Gustav Flink from the Greek σύγχΰσις “synchys” for “confounding” in allusion to its initially being mistaken for parisite.
Yellowish crystal of synchysite-(Ce) CaCe(CO3)2F on gneiss; Wanni glacier – Scherbadung area, Kriegalp Valley, Binn Valley, Wallis, Switzerland; FOV: 15 mm
Synchisite-(Ce) CaCe(CO3)2F crystal with honey color and evident cleavage on gneiss; Mt. Cervandone area (Scherbadung; Cherbadung), Devero Alp, Baceno, Devero Valley, Antigorio Valley, Ossola Valley, Verbano-Cusio-Ossola Province, Piedmont, Italy; crystal about 3 mm
Short prismatic and striated cristal of synchysite-(Ce) CaCe(CO3)2F with smoky quartz SiO2 on gneiss; Mt. Cervandone area (Scherbadung; Cherbadung), Devero Alp, Baceno, Devero Valley, Antigorio Valley, Ossola Valley, Verbano-Cusio-Ossola Province, Piedmont, Italy; quartz crystal: 7 mm
Orange-brown crystal of prismatic synchisite-(Ce) CaCe(CO3)2F on matrix; Wanni glacier – Scherbadung area, Kriegalp Valley, Binn Valley, Wallis, Switzerland; crystal 4 mm long
Prismatic elongated orange crystal of synchysite-(Ce) CaCe(CO3)2F on gneiss; Wanni glacier – Scherbadung area, Kriegalp Valley, Binn Valley, Wallis, Switzerland; crystal 4 mm long
A prismatic crystal of synchisite-(Ce) CaCe(CO3)2F, orange; Wanni glacier – Scherbadung area, Kriegalp Valley, Binn Valley, Wallis, Switzerland; crystal 4 mm long
Named after J.J. Paris, former Manager of the Muzo emerald mine, Muzo, Columbia (leasee of mine from 1828-1848).
Very rare; can be distinguished from Synchysite-(Ce) only by analytical methods.
Quite common on the market are pseudomorphs composed of earthy microporous muscovite aggregate with very minor admixture of earthy anatase from Mount Malosa, Zomba District, Malawi, sell as parisite (or pseudomoprhs after parisite). Recent investigation shows, that it is probably pseudomorphose after some silicate minerals (beryl, mylarite, cancrinite or something else).
Prismatic, striated pseudomorph after parisite-(Ce) CaCe2(CO3)3F2 with yellow-orange zircons ZrSiO4 and colorless quartz SiO2; Mount Malosa, Zomba District, Malawi; 26 x 21 x 13 mm; (two types of visually similar pseudomorphs are known from this locality a) type formed after REE-fluorcarbonates and always contain residual REEs and b) type formed after unknown hexagonal REE-free silicate and replaced by hydromuscovite with rutile admixture; it is rather imposible to recognize which one is this)
Cluster of lustrous, brown, elongated parisite-(Ce) CaCe2(CO3)3F2 crystals; Snowbird mine, Fish Creek, Alberton, Mineral Co., Montana, USA; 18 x 10 x 6 mm
Nice quality group of slender, deep reddish-brown prismatic parisite-(Ce) CaCe2(CO3)3F2 crystals; Snowbird mine, Fish Creek, Alberton, Mineral Co., Montana, USA; 18 x 10 x 6 mm
Prismatic and striated pale brown pseudomorph after crystal of parisite-(Ce) CaCe2(CO3)3F2 with yellow-orange zircon ZrSiO4 and colorless quartz SiO2; Mount Malosa, Zomba District, Malawi; 26 x 21 x 13 mm; (two types of visually similar pseudomorphs are known from this locality a) type formed after REE-fluorcarbonates and always contain residual REEs and b) type formed after unknown hexagonal REE-free silicate and replaced by hydromuscovite with rutile admixture; it is rather imposible to recognize which one is this)


-albite-quartz-czech-1")
-quartz-jasper-italy-1")


-albite-quartz-czech-3")

-quartz-jasper-italy-2")
-TL-belgium-3")
-TL-1")

-TL-belgium-4")
-quartz-jasper-italy-3")

-italy-2")

-TL-2")

![Group of acicular julgoldite-(Fe3+) Ca2Fe'''Fe'''2(Si2O7)(SiO4)O(OH)·H2O crystals on platy crystal of stilbite-Ca NaCa4[Al9Si27O72]·nH2O and chalcedony SiO2 matrix; Jalgaon Quarries, Jalgaon District, Maharashtra, India; 42 × 35 × 21 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/10/1093-julgoldite-fe3-stilbite-ca-chalcedony-india-2.jpg?w=209&h=210&ssl=1 "1093-julgoldite-(Fe3)-stilbite-Ca-chalcedony-india-2")
![Spectacular and very aesthetic group of julgoldite-(Fe3+) Ca2Fe'''Fe'''2(Si2O7)(SiO4)O(OH)·H2O crystals on platy crystal of stilbite-Ca NaCa4[Al9Si27O72]·nH2O and chalcedony SiO2 matrix; Jalgaon Quarries, Jalgaon District, Maharashtra, India; 42 × 35 × 21 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/10/1093-julgoldite-fe3-stilbite-ca-chalcedony-india-1.jpg?w=209&h=209&crop=1&ssl=1 "1093-julgoldite-(Fe3)-stilbite-Ca-chalcedony-india-1")

-stilbite-Ca-chalcedony-india-3")

![Dark Green acicular julgoldite-(Fe3+) Ca2Fe'''Fe'''2(Si2O7)(SiO4)O(OH)·H2O crystals inside and on the surface of platy crystal of stilbite-Ca NaCa4[Al9Si27O72]·nH2O; Jalgaon Quarries, Jalgaon District, Maharashtra, India; stilbite crystal: 33 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/10/1093-julgoldite-fe3-stilbite-ca-chalcedony-india-4.jpg?w=156&h=156&crop=1&ssl=1 "1093-julgoldite-(Fe3)-stilbite-Ca-chalcedony-india-4")












































































![Chunky fragment of alunite matrix with vugs filled with well crystallized reddish-brown lamellar alunite KAl3[(OH)6|(SO4)2]; Montioni, Suvereto, Livorno Province, Tuscany, Italy; 43 × 66 × 43 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/01/937-alunite-italy-3.jpg?w=423&h=423&crop=1&ssl=1 "937 alunite-italy-3")
![Alunite KAl3[(OH)6|(SO4)2] crystals on matrix from type locality; Allumiere Quarries, Allumiere, Tolfa Mts District, Rome Province, Latium, Italy; 40 × 23 × 20 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/02/1010-alunite-tl-italy-4.jpg?w=209&h=210&ssl=1 "1010-alunite-TL-italy-4")
![Minute reddish lustrous lamellar crystals of alunite KAl3[(OH)6|(SO4)2] on matrix, Montioni, Suvereto, Livorno Province, Tuscany, Italy; 43 × 66 × 43 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/01/937-alunite-italy-1.jpg?w=209&h=209&crop=1&ssl=1 "937 alunite-italy-1")
![White to brownish alunite KAl3[(OH)6|(SO4)2] crystals on flat matrix; Bomboly Hill, Mád, Zemplén Mts (Tokaj Mts), Borsod-Abaúj-Zemplén Co., Hungary; 27 × 15 × 12 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/02/1006-alunite-hungary-2.jpg?w=210&h=209&ssl=1 "1006-alunite-hungary-2")
![Crystal cluster of white to brownish (with inclusions of hematite Fe2O3), highly lustrous crystals of alunite KAl3[(OH)6|(SO4)2] to 5 mm; Bomboly Hill, Mád, Zemplén Mts (Tokaj Mts), Borsod-Abaúj-Zemplén Co., Hungary; 27 × 15 × 12 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/02/1006-alunite-hungary-1.jpg?w=209&h=209&crop=1&ssl=1 "1006-alunite-hungary-1")
![Orange-yellow alunite KAl3[(OH)6|(SO4)2] from type locality; Allumiere Quarries, Allumiere, Tolfa Mts District, Rome Province, Latium, Italy; 40 × 23 × 20 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/02/1010-alunite-tl-italy-2.jpg?w=209&h=209&crop=1&ssl=1 "1010-alunite-TL-italy-2")
![Orange-yellow, coloured with iron oxides alunite KAl3[(OH)6|(SO4)2] from type locality; Allumiere Quarries, Allumiere, Tolfa Mts District, Rome Province, Latium, Italy; 40 × 23 × 20 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/02/1010-alunite-tl-italy-1.jpg?w=156&h=156&crop=1&ssl=1 "1010-alunite-TL-italy-1")
![Sample covered by a layer of white to orange, prismatic alunite KAl3[(OH)6|(SO4)2] crystals from type locality; Allumiere Quarries, Allumiere, Tolfa Mts District, Rome Province, Latium, Italy; 40 × 23 × 20 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/02/1010-alunite-tl-italy-3.jpg?w=156&h=156&crop=1&ssl=1 "1010-alunite-TL-italy-3")
![Reddish lamellar crystals of alunite KAl3[(OH)6|(SO4)2] inside of the vugs, Montioni, Suvereto, Livorno Province, Tuscany, Italy; FOV: 30 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/01/937-alunite-italy-2.jpg?w=156&h=156&crop=1&ssl=1 "937 alunite-italy-2")
![White to brownish alunite KAl3[(OH)6|(SO4)2] cluster; Bomboly Hill, Mád, Zemplén Mts (Tokaj Mts), Borsod-Abaúj-Zemplén Co., Hungary; 27 × 15 × 12 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/02/1006-alunite-hungary-3.jpg?w=156&h=156&crop=1&ssl=1 "1006-alunite-hungary-3")
![Brownish jarosite KFe3[(OH)6|(SO4)2] 'rosettes' on goethite FeO(OH); Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; FOV: 15 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/01/862-jarosite-portugal-3.jpg?w=423&h=423&crop=1&ssl=1 "862-jarosite-portugal-3")
![Interesting combo specimen of light yellowish-brown crystals of natrojarosite NaFe3[(OH)6|(SO4)2] with brown crystals of jarosite KFe3[(OH)6|(SO4)2] on matrix; Christiana Mine, Kamariza Mines (Kamareza Mines), Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; 53 × 45 × 36 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1053-natrojarosite-jarosite-greece-3.jpg?w=209&h=210&ssl=1 "1053-natrojarosite-jarosite-greece-3")
![Cavity containing off-white masses of kamarizaite Fe3[(OH)3|(AsO4)2]*3H2O rimmed with brown jarosite KFe3[(OH)6|(SO4)2]; Kamariza Mines, Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; FOV: 30 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1033-kamarizaite-jarosite-greece-4.jpg?w=209&h=209&crop=1&ssl=1 "1033-kamarizaite-jarosite-greece-4")
![Rich specimen of kamarizaite Fe3[(OH)3|(AsO4)2]*3H2O present all around the specimen as off-white masses with small cavities with brown jarosite KFe3[(OH)6|(SO4)2]; Kamariza Mines, Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; 42 × 40 × 33 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1033-kamarizaite-jarosite-greece-1.jpg?w=210&h=209&ssl=1 "1033-kamarizaite-jarosite-greece-1")
![Light brown composite jarosite KFe3[(OH)6|(SO4)2] crystals on goethite FeO(OH); Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal;](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/01/862-jarosite-portugal-2.jpg?w=209&h=209&crop=1&ssl=1 "862-jarosite-portugal-2")
![Brown jarosite KFe3[(OH)6|(SO4)2] crystals richly scattered over goethitic matrix; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; 48 × 33 × 17 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1079-plumbojarosite-corkite-goethite-portugal-1.jpg?w=209&h=209&crop=1&ssl=1 "1079-plumbojarosite-corkite-goethite-portugal-1")
![Clusters of yellowish microcrystals of natrojarosite NaFe3[(OH)6|(SO4)2] with darker jarosite KFe3[(OH)6|(SO4)2] on matrix; Christiana Mine, Kamariza Mines (Kamareza Mines), Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; 53 × 45 × 36 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1053-natrojarosite-jarosite-greece-1.jpg?w=209&h=210&ssl=1 "1053-natrojarosite-jarosite-greece-1")
![Several light brown jarosite KFe3[(OH)6|(SO4)2] crystals on matrix; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; 39 × 26 × 21 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/01/862-jarosite-portugal-1.jpg?w=209&h=209&crop=1&ssl=1 "862-jarosite-portugal-1")

![Brown jarosite KFe3[(OH)6|(SO4)2] crystals richly scattered over matrix with tiny yellowish corkite PbFe3[(OH)6|SO4|PO4] (beudantite group) crystals on goethite FeO(OH) ; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; FOV: 20 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1079-plumbojarosite-corkite-goethite-portugal-2.jpg?w=316&h=316&crop=1&ssl=1 "1079-plumbojarosite-corkite-goethite-portugal-2")
![Light yellow plumbojarosite Pb0.5Fe3[(OH)6|(SO4)2] scatered over brown jarosite KFe3[(OH)6|(SO4)2] crystals in cavity; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; FOV: 35 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1079-plumbojarosite-corkite-goethite-portugal-4.jpg?w=316&h=316&crop=1&ssl=1 "1079-plumbojarosite-corkite-goethite-portugal-4")
![Yellowish-brown microcrystals of natrojarosite NaFe3[(OH)6|(SO4)2] with brown crystals of jarosite KFe3[(OH)6|(SO4)2] on matrix; Christiana Mine, Kamariza Mines (Kamareza Mines), Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; FOV: 20 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1053-natrojarosite-jarosite-greece-2.jpg?w=423&h=423&crop=1&ssl=1 "1053-natrojarosite-jarosite-greece-2")
![Superbly crystallized brownish jarosite KFe3[(OH)6|(SO4)2] with multiple twinnings richly scattered over matrix; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; FOV: 20 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1079-plumbojarosite-corkite-goethite-portugal-3.jpg?w=209&h=210&ssl=1 "1079-plumbojarosite-corkite-goethite-portugal-3")
![Sparkling, brown microcrystals of jarosite KFe3[(OH)6|(SO4)2] scattered throughout white sandstone matrix; Mercur District, (Camp Floyd District), Oquirrh Mts, Tooele Co., Utah, USA; 30 × 22 × 19 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1067-jarosite-usa-1.jpg?w=209&h=209&crop=1&ssl=1 "1067-jarosite-usa-1")
![Triangle vug filled with brown jarosite KFe3[(OH)6|(SO4)2] surrounded with whitish massive kamarizaite Fe3[(OH)3|(AsO4)2]*3H2O from type locality; Kamariza Mines, Agios Konstantinos, Lavrion District Mines, Lavrion District, Attikí Prefecture, Greece; vug 10 x 5 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1033-kamarizaite-jarosite-greece-2.jpg?w=210&h=209&ssl=1 "1033-kamarizaite-jarosite-greece-2")

![Light yellow plumbojarosite Pb0.5Fe3[(OH)6|(SO4)2] scatered over brown jarosite KFe3[(OH)6|(SO4)2] crystals on goethite FeO(OH) matrix; Serra da Mina Mine, Cercal, Santiago do Cacém, Setúbal District, Portugal; 48 × 33 × 17 mm](https://i0.wp.com/mineralcollectionblog.wordpress.com/wp-content/uploads/2018/03/1079-plumbojarosite-corkite-goethite-portugal-5.jpg?w=209&h=209&crop=1&ssl=1 "1079-plumbojarosite-corkite-goethite-portugal-5")
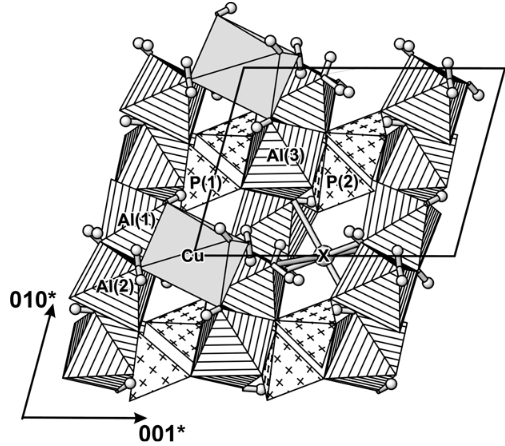
![Screenshot-2018-1-19 The turquoise a study of its history, mineralogy, geology, ethnology, archaeology, mythology, folkore,[...]](https://mineralcollectionblog.wordpress.com/wp-content/uploads/2018/01/screenshot-2018-1-19-the-turquoise-a-study-of-its-history-mineralogy-geology-ethnology-archaeology-mythology-folkore1.png)
















![TEM and electron diffraction study of (130) twin boundary in chrysoberyl from Pratinhas. (a) Bright-field TEM image of the twin boundary intersecting thin crystal part populated with oriented TiO2 precipitates. Electron diffraction pattern (EDP) recorded in domain I is rotated by ∼59.5° with respect to that of domain II, which exactly corresponds to the angle enclosed by the a-axes (marked by arrows), distinctive for (130) twin. (b) Twin boundary step to adjacent (130) planes. Note the moiré pattern in the transition area, produced by overlapping crystals in twin orientation. (c) Simulated EDP for (130) twin of chrysoberyl in [001] orientation based on crystallographic data obtained from Rietveld refinement (after Drev et al. 2105)](https://mineralcollectionblog.wordpress.com/wp-content/uploads/2017/11/figure-4-tem-and-electron-diffraction-study.png "Figure-4-TEM-and-electron-diffraction-study")






























































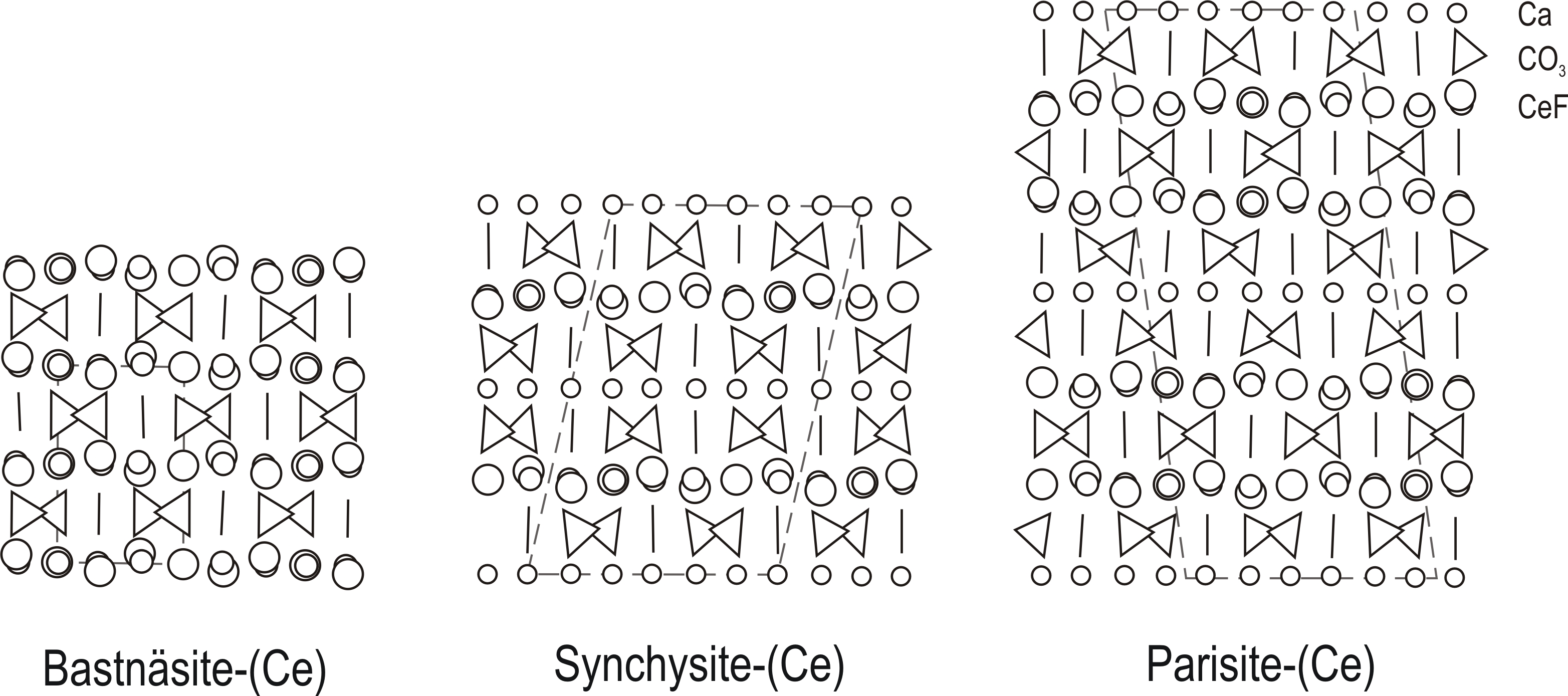
-1")

-2")
-2")
")












